Documente Academic
Documente Profesional
Documente Cultură
Tema IV
Încărcat de
Brice RodríguezTitlu original
Drepturi de autor
Formate disponibile
Partajați acest document
Partajați sau inserați document
Vi se pare util acest document?
Este necorespunzător acest conținut?
Raportați acest documentDrepturi de autor:
Formate disponibile
Tema IV
Încărcat de
Brice RodríguezDrepturi de autor:
Formate disponibile
GUA DE ESTUDIO
MODALIDAD ADI
TEMA No 4.
EQUILIBRIO DE FASES.
Profesora:
Ing. Koralys Goita
EQUILIBRIO DE FASES.
Muchas de las operaciones industriales como por ejemplo, la extraccin,
la absorcin, la destilacin o la adsorcin relacionan varias fases coexistiendo
en equilibrio. El equilibrio es una condicin esttica en la que no ocurren
cambios en las propiedades macroscpicas de un sistema con el tiempo; esto
implica un balance de todos los potenciales que podran causar un cambio. En
termodinmica un sistema en estado en equilibrio es aqul que se encuentra
en condiciones tales que no presentan ninguna tendencia para que ocurra un
cambio de estado.
La termodinmica del equilibrio de fases busca establecer las relaciones
entre varias propiedades. Si la temperatura, la presin y la concentracin de
las fases alcanzan sus valores finales, y en adelante permanecen constantes:
el sistema est en equilibrio, sin embargo, a nivel microscpico las condiciones
no son estticas; las molculas que se encuentran en una fase en un momento
dado no son las mismas en esa fase un instante despus. El flujo promedio de
las molculas es el mismo en ambas direcciones y no hay una transferencia
de materia neta entre las fases.
El tipo de problemas que intenta resolver la termodinmica de equilibrio
de fases se muestra en este tema para sistemas reales. Bsicamente
abordaremos para los sistemas reales, los clculos de composiciones en el
equilibrio, clculo de puntos de burbuja y puntos de roco.
Objetivo de la Unidad Temtica.
Capacitar al estudiante para que establezcan las desviaciones que representan
el sistema real respecto al equilibrio y tratamiento cualitativo en funcin del
conocimiento de la T, P y composicin del sistema.
4.1. REGLA DE LA FASES.
Para un sistema heterogneo compuesto por n fases y m componentes
se cumple que:
(4.1)
I
(1)
= I
(2)
= . . = I
(n)
P
(1)
= P
(2)
= . . = P
(n)
p
1
(1)
= p
1
(2)
= . . = p
1
(n)
p
2
(1)
.
.
=
p
2
(2)
.
.
= . . =
p
2
(n)
.
.
p
m
(1)
= p
m
(2)
= . . = p
m
(n)
m
+2)
m
+ 1)
m + 1)
m + 1)
m + 1)
equilibrio interno, entonces hay que tener en cuenta que de las n(m + 1)
En el sistema expuesto en (4.1) podemos interpretar, el equilibrio
trmico, dada la igualdad de temperaturas en las diferentes fases, el equilibrio
mecnico, dado por la igualdad de presin en todas las fases coexistentes, y el
equilibrio de fases, dado por la igualdad de potenciales qumicos en todas las
fases.
Se puede caracterizar el estado intensivo de cada fase presente en un
sistema heterogneo en equilibrio interno por la T, P, y p de cada
componente presente, es decir un total de (m variables (m. las sustancias
presentes y 2, de la T y P). Sin embargo no todas estas variables son
independientes, la ecuacin ya estudiada de Gibbs Duhem, establece cuantas
variables estn relacionadas. La ecuacin de Gibbs Duhem (ecuacin 3.15 del
tema 3) representa una restriccin en la variacin simultnea de T, P, y p , de
una misma fase. Por consiguiente de las variables intensivas que pueden
usarse para caracterizar una fase, slo (m son variables independientes; es
decir una fase tiene ( grados de libertad.
Si el sistema heterogneo no se encuentra en un estado de equilibrio
interno, pero cada fase lo est, el nmero de variables es n( , por cada
fase tiene ( grados de libertad. Cuando el sistema est en estado de
variables hay (n - 1)(m + 2) relaciones de equilibrio representadas
ecuaciones (4.1). Los grados de libertad ser entonces el nmero de variables
intensivas utilizadas para caracterizar el sistema menos el nmero de
restricciones o relaciones entre ellas.
F = N
uubIcs ntcn
por las
sus
-N
cIuconcs
F = n m + 1) - (n - 1)(m + 2)
F = m +2 - n (4.2)
La ecuacin (4.2) es la ecuacin de la e la fase para sistemas sin
ROBLEMA 1
) Calcular el nmero de grados de libertad que definen un sistema
olucin
de componentes = nmero de especies qumicas diferentes, en este
l nmero de fases = tendremos dos fases, la disolucin de sacarosa que ser
o existen ni reacciones ni relaciones entre los componentes por lo tanto el
s decir con dos variables independientes podremos definir el sistema, estas
(
regla d
reaccin qumica.
P
a
compuesto por sacarosa slida en equilibrio con una disolucin
acuosa de sacarosa.
S
El nmero
caso ser 2, la sacarosa y el agua. Por lo tanto m = 2
E
una fase lquida y la sacarosa slida. Por lo tanto n = 2
N
nmero de grados de libertad ser: F=2-2+2=2
E
dos variables pueden ser por ejemplo la presin y la temperatura, ya que a
una (P,T) dada la solubilidad de la sacarosa slo tiene un valor posible, y es el
que determina la concentracin de sacarosa en agua.
b) Calcular el nmero de grados de libertad para una mezcla gaseosa
de N
2
, H
2
y NH
3
que no reaccionan entre s.
Solucin
m = 3, las tres especies qumicas diferentes.
n = 1 una nica fase gaseosa
No existen relaciones entre los componentes del sistema, luego F=3-1+2=4
4.2. COMPORTAMIENTO CUALITATIVO DE LAS FASES PARA SISTEMAS
LQUIDO-VAPOR.
Para estudiar de modo cualitativo el comportamiento de las fases
coexistiendo (lquido y vapor), haremos uso de los diagramas estudiados en la
seccin siguiente, lo cual nos permitir, entender con mayor claridad, los
fenmenos que ocurren.
En esta presentacin cualitativa, el estudio se va a limitar a sistemas
constituidos por dos compuestos qumicos, puesto que los sistemas de mayor
complejidad no pueden ser representados grficamente en forma adecuada.
Cuando m=2 la regla de la fases queda para un sistema lquido vapor
como: F=2. Entonces podramos representar en un diagrama PTxy el equilibrio
lquido vapor. En la Fig. 4.1 se representa este tipo de diagrama de manera
tridimensional.
4.3. DIAGRAMAS DE EQUILIBRIO:
En el libro gua, (Smith y Col.) se encuentra este apartado, desarrollado
a mayor profundidad, la intencin de esta gua es esbozar las ideas ms
resaltantes del estudio cualitativo de las fases lquido vapor en coexistencia. El
diagrama PTxy representado en la Fig. 4.1, tiene un grado de complejidad
mayor a los estudiados anteriormente con dos dimensiones. El diagrama de la
Fig. 4.1, representa el espacio tridimensional de P-T y la composicin de un
sistema binario. Los puntos C
1
C
2
, representan los puntos crticos para cada
una de las sustancias en estado puro. La superficie P-T-y
1
, incluye los estados
de vapor saturado, y la superficie P-T-x
1
, los estados de lquido saturado.
Fig. 4.1. Diagrama tridimensional PTxy para el equilibrio lquido/ vapor.
Fuente: Smith. Sptima edicin.
La regin donde est situado el punto F, es lquido subenfriado, si se
reduce la P a T y composicin constante, se traza una lnea vertical FG, la
primera burbuja de vapor aparece en el punto L. Conforme sigue en descenso
la P, se vaporiza en mayor cantidad el lquido hasta W, que representa el
estado de vapor saturado.
Fig. 4.2. a) Diagrama Pxy para tres temperaturas. b) Diagrama Txy para tres
presiones. ) IiquiJo soturoJo (linco Jc burbu]o) - - - Iopor soturoJo (linco Jc rocio
Fuente: Smith. Sptima edicin
Un plano horizontal que atraviesa la fig. 4.1 y es perpendicular al eje P,
se identifica por KJIHLK. Las lneas sobre este plano expresan un diagrama T-
xy. Proyectndolas para varias presiones, el diagrama resultante es el 4.2.b.,
similar a la 4.2.a, pero para tres diferentes presiones.
Fig.4.3. Parte de un Diagrama PT en la regin crtica.
Fuente: Smith. Sptima edicin
En la Fig. 4.3, el punto C significa el punto crtico. Las curvas
discontinuas del sistema indican la fraccin global del sistema. A la izquierda
de C, una reduccin en la P a lo largo de la lnea BD, se acompaa por la
vaporizacin de lquido del punto de burbuja al punto de roco. La condicin
original es el punto F, un estado de vapor saturado, la licuefaccin se suscita
hasta por la reduccin de P y logra un mximo en el punto G, despus del cual
la vaporizacin tiene lugar hasta que se logra el punto de roco en el punto H.
Esto se conoce como condensacin retrgrada. Esto se produce cuando un
gas a alta presin se expande luego de disolver algo lquido ya que pierde su
capacidad de disolver lquidos comportndose como un gas con las molculas
distanciadas y los componentes pesados se desprenden generando la
condensacin retrgrada. Lo mismo ocurre cuando un gas a alta presin se
calienta (en forma isobrica). La expansin trmica aleja las molculas del gas
y los componentes pesados se desprenden de la masa gaseosa.
Fig. 4.4. Caminos termodinmicos para la misma mezcla.
Fuente: M. Crotti, S. Bosco
En la Fig. 4.4. observamos una mezcla que se encuentra en la condicin
inicial X, el camino a, representa una despresurizacin isotrmica, y el camino
b, un calentamiento isobrico.
Camino "a". Los ingenieros de petrleo califican este tipo de mezcla X
como un Petrleo muy subsaturado. Cuando la presin desciende lo suficiente
se comienza a liberar gas en el seno del fluido, dado que se "corta" la campana
por la curva de presiones de burbuja (Trazo azul).
Camino "b". Es un camino termodinmico perfectamente vlido, pero
infrecuente en la industria del petrleo. Lo interesante de este camino es que
cuando la temperatura sube lo suficiente, del seno de la muestra comienza a
liberarse lquido (se "corta" la campana por la curva de presiones de roco
representada con trazo rojo). De este modo calificaramos a la mezcla en el
punto "X" como Gas.
Fig. 4.5. Diagrama yx para el Etano/n-heptano.
Fuente: Smith. Sptima edicin
En la Fig. 4.5, se observa un diagrama y
1
-x
1
, para varias presiones del
mismo sistema. De acuerdo a la convencin, las fracciones mol de las especies
ms voltiles de la mezcla se grafican como y
1
y x
1
. Las concentraciones
mximas y mnimas de las especies ms voltiles que se obtienen por
destilacin a una presin conocida se sealan por los puntos de interseccin de
la curva y1-x1 apropiada con la diagonal, a causa de tales puntos el vapor y el
lquido tiene la misma composicin El punto A representa la composicin de las
fases de vapor y lquido a la presin mxima a la cual estas coexisten.
(Tomado de: Smith y col.).
4.4. COMPORTAMIENTO AZEOTRPICO
Como se estudio anteriormente en la seccin 3.9, del tema 3, existen
gran cantidad de soluciones con comportamiento real. Otro de los casos de
mezcla de soluciones con comportamiento real es cuando se presentan los
llamados azetropos, en lo cual el aspecto ms resaltante de las curvas de
presin total en sistemas con desviaciones positivas de la ley de Raoult,
presentan un punto mximo, donde la composicin de la fase lquida es
igual a la composicin de la fase vapor. Es decir, los azetropos son
sistemas formados por 2 o ms sustancias, en el cual la composicin de la fase
lquida iguala a la fase gaseosa:
x
1
= y
1
(4.3)
La fig. 4.6, representa un azetropo con desviaciones positivas de la
linealidad (de Raoult) lo bastantes grandes como para causar mximos en la
curva P-x
1
. Este es el caso de un azetropo de presin mxima. En el cual
un lquido en ebullicin con una composicin x
1
produce un vapor de idntica
composicin y
1
. En este estado el lquido no cambia su composicin conforme
se evapora, por lo que no es posible la separacin por destilacin de tal
solucin de ebullicin constante.
Fig. 4.6. Etano (1)/tolueno(2) a 65C. Lnea discontinua relacin Px para la
ley de Raoult. Fuente: Smith. Sptima edicin
Fig. 4.7. Cloroformo (1)/tetrahidrofurano(2) a 30C. Lnea discontinua
relacin Px para la ley de Raoult. Fuente: Smith. Sptima edicin
La Fig. 4.7, representa una desviacin negativa de la linealidad, y es lo
suficientemente grande con respecto a las diferencias entre las presiones de
vapor de las especies puras, la curva P-x presenta un mnimo. La curva P-y
es tangente a la P-x donde se presenta en azetropo.
Fig. 4.8. Etanol (1)/Tolueno(2) a 1 amt. Fuente: Smith. Sptima edicin
Fig. 4.9. Cloroformo (1)/Tetracloruro de carbono(2) a 1 amt. Fuente: Smith.
Sptima edicin
En la Fig. 4.8, se representa un azetropo de punto de ebullicin
mnimo, hierve a temperatura menor que cualquiera de los componentes
puros. Cuando se destila un sistema de estos componentes, el producto del
tope es el azetropo.
En la Fig. 4.9, se representa un azetropo de punto de ebullicin
mximo hierve a temperatura mayor que la de los componentes puros y por lo
tanto siempre sale por el fondo de la columna.
Determinacin cuantitativa de Azetropo.
Para determinar cuantitativamente la presencia de un azetropo, es
necesario definir un nuevo concepto que nos permitir realizar el clculo. Este
concepto se le llama volatilidad relativa, que no es ms que la relacin
existente entre las contantes de equilibrio.
o
]
=
K
K
]
(4.4)
P y
= y
sut
x
(S.11u)
P = y
sut
(4.6)
y
Azc
=
P
Azc
P
sut
En el equilibrio sabemos que de acuerdo a la ecuacin (3.51) estudiada
en el tema 3, podemos escribir entonces para un sistema lquido-vapor:
l
i
v
i
f f
=
(4.5)
Para un sistema como fase lquida real, y fase de vapor ideal,
recordando la ecuacin del tema 3 (3.110) obtenemos la ecuacin de
equilibrio para este sistema.
En el azetropo, tanto para la ecuacin (3.110) y (3.108) se cumple la
ecuacin (4.3) por lo tanto para la ecuacin (3.110) queda:
Entonces, el coeficiente de actividad en el azetropo puede ser
determinado mediante la ecuacin (4.6).
(4.7)
y
Azc
=
P
Azc
=
Coeficiente de actividad del componente i en el azetropo
Presin del sistema en el azetropo.
Como en el azetropo se cumple la ecuacin (4.3), y de acuerdo a la
definicin de la constante de equilibrio estudiada en el tema 2 apartado 2.5, la
volatilidad relativa en este estado azetropico es:
o
]
=
K
K
]
(4.8)
o
]
=
y
y
]
x
]
= 1 (4.9)
K
=
y
sut
P
Si = 1, no hay separacin entre los componentes i y j. En el azetropo se
cumple la ecuacin (4.9).
ij
La constante de equilibrio para la ecuacin (3.110) puede definirse como:
(4.1u)
o
]
=
y
sut
P
Sustituyendo (4.10) en la ecuacin (4.8), se tiene:
y
]
P
]
sut
P
(4.11)
o
]
=
y
sut
y
]
P
]
sut
Simplificando la ecuacin (4.11), queda:
(4.12)
o
12
=
y
1
P
1
sut
y
2
P
2
sut
La ecuacin (4.12) se evala a diluciones infinitas, es decir cuando x
1
=0 y
x
2
=0 ( x
1
=1).
Supongamos para una mezcla binaria:
(4.1S)
1
2
= 1
o
12
=
y
1
P
1
sut
P
2
sut
Cuando x
1
=0 entonces y y y como se explico en el problema 3 de la
gua de estudio del tema 3. La ecuacin (4.13) que:
(4.14)
2
1
= 1
o
12
=
P
1
sut
y
2
P
2
sut
Cuando x
2
=0 entonces y y y
(4.1S)
Es decir, con la ecuacin (4.14) y (4.15) se evalan los extremos de las
composiciones.
PROBLEMA 2
Verificar si el sistema 2,4 dimetil pentano(1)-benceno(2) forma un
azetropo a 60 C.
Las presiones de vapor son:
o
P
1
= 51.7 kPa ; = 51.6 kPa
o
P
2
Los coeficientes de actividad a diluciones infinitas son:
1
= 1.96 ; = 1.48
Solucin
x
1
0
12
= 1.96x51.7/51.6 = 1.96
x
1
1
12
= 51.5/1.48x51.6 = 0.68
Ya que
12
es una funcin continua de x
1
, debe haber un valor de x
1
para el
cual
12
= 1 (azetropo de presin mxima).
Los y y y son datos especificados en este problema, sin embargo cuando
no han sido reportados, deben ser calculados por alguno de los modelos de
solucin como el problema 9 del tema 3. Con el simulador PRO-II, podemos
obtener los coeficientes de actividad para este sistema, pero como no se ha
especificado el modelo de solucin a seguir, se debe tantear cul de los
modelos de solucin reporta con mejor cercana los coeficientes a diluciones
infinitas reportados en este problema. De este modo obtendremos, la grfica
del azetropo. En la Fig. 4.10 podemos observarla. El modelo que se ajusto
con ms exactitud fue UNIQUAC. En la Fig. 4.11 podemos observar los
coeficientes de actividad a diferentes composiciones y los de dilucin infinita.
1
Composition, Mole Fraction 24DMP, (T = 60.000 C)
0 0.2 0.4 0.6 0.8 1.0
P
r
e
s
s
u
r
e
,
k
P
a
52.0
54.0
56.0
58.0
60.0
P-X-Y Plot for 24DMP and BENZENE
B Bubble Point
B
B
B
B
B
B
B
B
B B
B
B
B
B
B
B
B
B
B
B
B
D DewPoi nt
D
D
D
D
D
D
D
D
D D
D
D
D
D
D
D
D
D
D
D
D
Fig. 4.10. Diagrama de Equilibrio P-xy del sistema 2,4 dimetilpentano(1)-
benceno(2) a 60C. Fuente: Propia, elaborada en simulador PRO-II.
Liquid Composition, Mole Fraction 24DMP
0 0.2 0.4 0.6 0.8 1.0
A
c
t
i
v
i
t
y
C
o
e
f
f
i
c
i
e
n
t
0.90
1.20
1.50
1.80
2.10
Liquid Activity Plot for 24DMP and BENZENE
1 24DMP
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1 1 1 1 1
2 BENZENE
2 2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
2
Fig. 4.11. Coeficiente de actividad para sistema 2,4 dimetilpentano(1)-
benceno(2) a 60C. Fuente: Propia, elaborada en simulador PRO-II.
Liquid Composition, Mole Fraction 24DMP
0 0.2 0.4 0.6 0.8 1.0
V
a
p
o
r
C
o
m
p
o
s
i
t
i
o
n
,
M
o
l
e
F
r
a
c
t
i
o
n
2
4
D
M
P
0
0.2
0.4
0.6
0.8
1.0
X-Y Plot for 24DMP and BENZENE
x = y
Equi libr iumcurv e
Fig. 4.12. Diagrama de Equilibrio x-y del sistema 2,4 dimetilpentano(1)-
benceno(2) a 60C. Fuente: Propia, elaborada en simulador PRO-II.
En los sistemas con grandes desviaciones de la idealidad y/o puntos de
ebullicin prximos de los componentes puros a menudo dan lugar a la
formacin de azetropo.
En la fig. 4.12 podemos observar en el diagrama y-x como existe un punto de
interseccin de la curva de equilibrio con la diagonal de 45, esto muestra la
presencia del azetropo.
4.5. FUGACIDAD EN FUNCIN DE LA COMPOSICIN. LEY DE HENRY Y
LA CONCEPCIN DE LEWIA-RANDALL DEL MODELO DE RAOULT.
En el tema 3, estudiamos la definicin del coeficiente de actividad, y se defini
en la ecuacin (3.77) como:
y
=
`
(S.77)
= 1 Cuando y decimos que la fugacidad del componente i en solucin es:
=
y
=
`
0
x
(4.16)
es la fugacidad del liquido puro a la T, y P de la mezcla.
La ecuacin (4.16) es conocida como la ecuacin de Lewis Randall, cuando xi
tiende a la unidad, la fugacidad de i es igual a la fugacidad del componente
puro.
Las ecuaciones, (3.77) y (4.16), pueden generalizarse para otros estados de
referencia, es decir:
(4.17)
0
x
0
=
`
(4.18)
fugacidad del estado estndar seleccionado, a la T, y P de la mezcla.
Cuando se fija otro estado de referencia, aparece otra ecuacin que tambin
expresa la proporcionalidad directa entre la
y xi. Esto se hace cuando la
sustancia pura no existe como lquido a las condiciones de T y P de la mezcla,
por esta razn debe ser tomado otro estado de referencia. Esto se aplica con
bastante frecuencia cuando gases y slidos de solubilidades limitadas se
disuelven en lquido.
H
i
f
i
Fig. 4.3 Dependencia con la composicin de las fugacidades de fase lquida
para las especies i en una solucin binaria. Fuente: Smith. Sptima edicin
Cuando xi tiende a cero, la lnea en la Fig. 4.13 que representa el estado de
referencia ms cercano al real viene expresada matemticamente de la
tangencia:
lim
x
i--
o
`
= K
(4.19)
= K
=
|
=
|
|
= K
|
y
(L.H)
=
`
La ecuacin (4.18) la escribimos de la siguiente manera:
(4.20)
La ecuacin (4.20) tiene validez cuando los valores de xi son muy pequeos.
factor de proporcionalidad, constante de Henry.
Estado de referencia para Lewis Randall:
Estado de referencia para Ley de Henry:
Estas ecuaciones representan (ambos modelos de idealidad) dos usos
importantes: 1) Da valores aproximados de las fugacidades en solucin cuando
se acerca a intervalos apropiados de composicin, el 2) Provee valores de
referencia para comparar los valores de las fugacidades en solucin.
Entonces, para Henry tenemos que la ecuacin (4.17) se convierte en:
(4.21)
y
=
`
Tambin sabemos que:
(S.77)
=
`
De la ecuacin (3.77) tenemos que:
(4.22)
lim
x
i-
De la ecuacin (4.19) tenemos:
-
o
`
= K
(4.19)
K
(4.2S)
Como xi tiende a cero, entonces sustituyendo (4.19) en (4.22)
Despejamos de (4.21) y (3.77) , e igualamos:
`
= y
(L.H)
K
y
(L.H)
=
y
(4.24)
y
(L.H)
=
y
Sustituimos (4.23) en (4.24) y tenemos:
(4.2S)
La ecuacin (4.25) nos permite calcular el coeficiente de actividad para la ley
de Henry a partir, de los coeficientes de actividad basados en la regla de
Lewis/Randall, calculados por modelos de solucin.
PROBLEMA 3
La fugacidad del componente 1 en una mezcla lquida binaria a 298K y 20bar
viene dada por:
3
1
2
1 1 1
40 80 50 x x x f + =
Donde la fugacidad esta en bar y x
1
es la fraccin molar. Determine:
(a) La fugacidad del componente puro 1.
(b) El coeficiente de fugacidad
1
(c) La constante de Henry K
1
(d) El coeficiente de actividad.
Solucin
(a) Cuando x
1
=1 entonces:
bar f 10 40 80 50
1
= + =
(b)
5 . 0
20
10
1
1
= = =
P
f
r
K
1
= lim
x
i-
(c) La constante de Henry se
o
`
1
x
1 -
K
1
= lim
x
i--
o
(Su - 8u x
1
+ 4u x
1
2
) = Su bor
2
1 1
1
3
1
2
1 1
1 1
1
1
8 5
10
40 80 50
x x
x
x x x
f x
f
+ =
+
= =
v
-
L
(d)
4
4.6. LA CONSTANTE DE EQUILIBRIO.
Como se estudio en apartados anteriores en el tema 2, la constante de
equilibrio, se determina de acuerdo al tipo del sistema en estudio. Para abarcar
a mayor profundidad las diferentes constantes de equilibrio, profundizaremos
los mtodos termodinmicos que se utilizan. En la tabla 3.8, del tema 3, se
introdujo a dicho estudio, en donde se menciono a groso modo las ecuaciones
de equilibrio para cada mtodo. Todas las ecuaciones que desarrollaran estn
basadas en el criterio de equilibrio visto en el tema 3 en la ecuacin (3.105)
para el equilibrio lquido-vapor:
l
i
v
i
f f
=
(3.105)
Desarrollemos a partir de la ecuacin (3.105) las diferentes constantes
de equilibrio. Para estudiarla con mayor claridad dividamos en 2 grandes
grupos:
1) Mtodo de ecuaciones de estado, conocido como EOS (equations of
state).
Tambin conocido como mtodo , en este mtodo tanto la fase lquida
como la fase de vapor se ajusta a una ecuacin de estado para todo el
intervalo de presiones. En este mtodo podemos encontrarnos como varios
casos:
Caso A: Fase de vapor real- fase lquida real.
La ecuacin (3.106) vista en el tema 3 tenemos la fugacidad para la fase
gaseosa:
=
i
v
P y
(S.1u6)
i
L
=
i
L
P x
(4.26)
v
y
=
i
L
P x
(4.27)
K
=
i
v
Del mismo modo podemos escribir la fugacidad para la fase lquida como:
Igualando de acuerdo a (3.105), se obtiene que:
P
Despejando la constante de equilibrio:
(4.28)
P y
= y
(S.1u7o)
K
=
y
i
P
Donde los coeficientes de fugacidades de ambas fases son funciones de T, P y
composicin de cada una de las fases. Es por ello que los clculos de
vaporizaciones instantneas, suelen ser procesos iterativos, ya que se
desconocen las composiciones, y las constantes de equilibrio son un funcin de
las mismas. La ecuacin (4.28) representa la No-idealidad de ambas fases.
2) Mtodos de modelos de solucin. Conocido como
- y
Caso B: Fase de vapor real- fase lquida real.
Este caso ya ha sido estudiado en el tema 3, la ecuacin de equilibrio es:
La constante de equilibrio es:
(4.29)
|
=
|
el estado estndar seleccionado es el del lquido puro a T y P de
la mezcla. En la ecuacin (4.29) podemos observar la dependencia de la T, P,
y composicin de ambas fases. Para resolver problemas de equilibrio de fases,
los clculos son iterativos, y normalmente los podemos resolver mediante la
ayuda de un simulador de procesos.
Caso C: Fase de vapor ideal- fase lquida real.
De la ecuacin (3.107a), simplificamos algunos trminos como se hizo en tema
3, y se estable la ecuacin ya estudiada:
P y
= y
sut
x
(S.11u)
K
=
y
sut
P
La constante de equilibrio queda entonces como:
(4.Su)
P y
= P
sut
x
K
=
P
sut
P
La ecuacin (4.30) es funcin de la composicin de la fase lquida, la T y P. La
, es en el sentido de la ley de Raoult, es decir que el estado estndar
seleccionado es el del lquido puro a T y P de la mezcla.
Caso D: Fase de vapor ideal- fase lquida ideal.
Este es el caso ms sencillo, la constante de equilibrio no depende de la
composicin. Este es el caso estudiado en el tema 2, la ley de Raoutl, la
ecuacin de equilibrio queda simplificando el coeficiente de actividad de la
ecuacin (3.110) como:
Ley de Raoult.
La constante queda
(4.S1)
Caso E: Estado estndar seleccionado, constante de Henry.
Ya que el estado de referencia es H
i
, la ecuacin de equilibrio para un sistema
de ambas fases reales es:
i
P y
= y
(4.S2)
K
=
y
(L.H)
E
i
P
La ecuacin (4.29) se convierte en:
(4.SS)
K
=
y
(L.H)
E
P
La ecuacin (4.30) se convierte en:
(4.S4)
K
=
E
P
La ecuacin (4.31) queda:
(4.SS)
y
(L.H)
K
=
y
v
P
: se calcula por (4.25)
Mtodos para determinar la constante de equilibrio.
Existen diferentes mtodos para calcular la constante de equilibrio, basados en
los casos estudiados con anterioridad. En este apartado estudiaremos 2, el
Mtodo de Chao-Seader, y el mtodo del Nomograma de DePriester.
Mtodo de Chao Seader
Para las especies menos voltiles de una mezcla, la dependencia de los
valores de K con respecto a la composicin se debe principalmente al
comportamiento no ideal (Seader y Col.). Este mtodo est basado en que la
no idealidad es debida a diferencias de las fuerzas de Van del Waals de
atraccin entre las especies presentes. Las soluciones de mezclado tiene un
calor de mezclado endotrmico y todos los coeficientes de actividad son
mayores a la unidad. (Profundizar en texto Operaciones de separacin por
etapas de equilibrio en ingeniera qumica de Seader y col.).
La constante de equilibrio para este mtodo est basada en la ecuacin
(4.29) ya estudiada:
(4.29)
Donde, debido a la estrecha relacin entre la fugacidad y presin, se define un
cociente para sustancias puras:
L
=
0
P
(4.S6)
0
=
K
=
y
v
Donde es el coeficiente de fugacidad de la especie lquida pura y
es
la fugacidad del lquido puro.
(4.S7)
1
]
=
x
]
:
]L
x
C
=1
:
L
La constante de equilibrio la podemos escribir entonces como:
Asumiendo volmenes molares aditivos queda:
=
x
]
:
]L
:
L
(4.S8)
1 =
Donde:
Fraccin en volumen, suponiendo volmenes molares aditivos. Las
composiciones deben ser v/v.
Para los coeficientes de fugacidad en la fase vapor utiliz la ecuacin de
Redlich-Kwong, y para los coeficientes de actividad utiliz la ecuacin de las
soluciones regulares:
RT
v
C
i
j i iL
i
2
1
j
) (
ln
=
=
(4.39)
Donde:
o
) 1 (
,
) 0 (
, ,
log log log
L i i L i
o
L i
w + =
= parmetro de solubilidad, normalmente se toman como constante. En el
anexo 4.1, se pueden observar algunos parmetros estimados para esta
correlacin.
Chao y Seader desarrollaron una ecuacin emprica para en funcin de
Tr, Pr y w, basado en el principio de los estados correspondientes (P = ZRT/v, Z
= Z(Pr,Tr,w)):
(4.40)
Donde, los parmetros de la ecuacin 4.40 se determina por los mostrados en la
Fig.4.4.
Fig. 4.4. Parmetros para calcular el coeficiente de fugacidad puro.
Fuente: Seader y col.
La ecuacin de Chao y Seader se utiliza ampliamente en la industria
petrolera y del gas natural para el clculo de K en parafinas, olefinas,
aromticos, naftenos e hidrgeno, siguiendo las siguientes restricciones:
a) T < 260 C
b) P < 6.9 MPa
c) Para hidrocarburos (excepto metano), 0.5 < Tr
i
< 1.3 y presin crtica de la
mezcla < 0.8.
d) Para sistemas conteniendo metano y/o hidrgeno, la Tr molar promedio <
0.93, y la fraccin molar de metano 0.3. La fraccin molar de otros gases
disueltos < 0.2
e) Cuando se calcula K de parafinas u olefinas, la fraccin de los aromticos en
la fase lquida debe ser < 0.5; y para la K de aromticos, la fraccin molar de la
fase lquida aromtica debe ser > 0.5
PROBLEMA 4
Estmese el valor de la constante de equilibrio para el benceno, en una
solucin con n-propano a 477,59 K y presin de 2,829 MPa. Utilice el
Mtodo de Chao-Seader. El propano tiene un porcentaje peso de
26,92% en la fase de vapor y 6.46% en la fase lquida.
Solucin
Para la resolucin de este ejercicio, utilizaremos una herramienta muy til, el
programa Mathcad. Y corroboraremos el resultado con lo reportado por la
literatura.
Propiedades crticas para el n-propano y el benceno.
Tabla 4.1. Propiedades Crticas.
Fuente: Smith. Sptima edicin
Tc (K) Pc (kPa)
Vc
(m
3
/kgmol) Zc w
369,8 4248 0,2 0,276 0,152
562,2 4898 0,259 0,271 0,21
Las composiciones fueron dadas en %p/p, debemos hacer la transformacin a
concentracin molar. En la tabla 4.2 y 4.3, se observan estos valores.
Tabla 4.2. Fracciones molares de los componentes en fase vapor.
Componente kg PM kgmol yi
1 (n-C
3
) 26,92 44,097 0,6105 0,395
2 (C
6
) 73,08 78,114 0,9356 0,605
Suma 1,546 1
Fuente: Propia
Tabla 4.3. Fracciones molares de los componentes en fase lquida.
Componente kg PM kgmol xi
1 (n-C
3
) 6,46 44,097 0,1465 0,1090
2 (C
6
) 93,54 78,114 1,1975 0,8910
Suma 1,3440 1,0000
Fuente: Propia
La constante de equilibrio experimental es:
K
2
=
y
2
x
2
K
2
= u,67916
K
=
y
v
De la ecuacin (4.37) la constante de equilibrio ser calculada por:
(4.S7)
t
V
Debemos calcular cada uno de los parmetros que aparecen en la ecuacin
(4.37) para determinar la constante de equilibrio para el sistema en estudio.
Clculo del coeficiente de fugacidad en la mezcla, de vapor q .
El mtodo de Chao-Seader, establece que el coeficiente de fugacidad de la fase
de vapor se calcula por la ecuacin de Redlich-Kwong. Para ello utilizamos
las ecuaciones del formulario del tema 3, el apartado 3, estudia de modo
detallado esta ecuacin. Utilizamos la ecuacin (f.3.20), para calcular el
coeficiente de fugacidad de la fase gaseosa.
( )
z
B z
b
b
B
A
B z z
b
b
i
i i
i
2
2 2
ln ) ln( 1
ln
*
*
*
*
+
+ == (f.3.20)
Se deben calcular todos los parmetros que aparecen en la ecuacin (f.3.20).
Los parmetros A
*
, B
*
, a y b, deben ser calculados por regla de mezcla con las
ecuaciones (f.3.18) que aparecen en el mismo apartado:
( )
ji ij i i
ij j i ij j i ij
k k b Y b
a Y Y a k a a a
= =
= =
1
(f.3.18)
Para ellos debemos calcular los parmetros de los componentes puros con las
ecuaciones:
Pc
Tc R
b
T Pc
a
i
i
=
=
08664 , 0
5 . 0
o
1
= 87S,uS
k[ m
3
Kgmol
2
Tc R 42748 , 0
5 , 2 2
(f.3.15)
Los parmetros resultantes de (f.3.15) son:
o
2
= 8,79
k[ m
3
Kgmol
2
2u6
b
1
= u,u6S
m
3
Kgmol
b = u,u8S
m
3
Kgmol
2
o = 1S16,77
k[ m
3
Kgmol
2
Como se desconoce K
ij
se asume cero (0). Aplicando (f.3.18) resulta:
b = u,u748
m
3
Kgmol
Se calculan los parmetros A
*
y B
*
con (f.3.17):
272 , 0
2 2
*
= =
T R
aP
A 053 , 0
*
= =
RT
bP
B
Falta determinar el parmetro z, que aparece en (f.3.20). Para ello resolvemos
el polinomio cbico con (f.3.16):
( ) 0
* * 2 * * * 2 3
= + B A Z B B A Z Z
Para resolver este polinomio utilizamos la herramienta mathcad, como
podemos observar en la fig.4.5.
3 races arrojadas por programa
Fig. 4.5 Pantalla del Programa Mathcad. Fuente: Propia.
En la Fig. 4.5 podemos observar que la resolucin del polinomio, trae
como resultado 3 races, matemticamente este resultado es obvio.
Termodinmicamente debemos determinar cul de las 3 races es la correcta
para resolver este ejercicio.
Para ello analicemos la siguiente Fig.4.6, donde podemos observar como
el lazo (isoterma) de la ecuacin de orden 3, toca el estado de lquido saturado
y el de vapor saturado, dando valores exactos en estos dos estados
termodinmicos.
Fig. 4.6 Isoterma proporcionadas por ecuaciones cbicas de estado.
Fuente: M. Fidel 2005.
Los dos posibles resultados para 3 races del V para cada (P,T) :
Dos races significativas en la zona de dos fases (mayor: para el vapor y
intermedia: no significativa, menor: para el lquido).
Una sola raz significativa en las zonas monofsicas.
En la Fig. 4.5 podemos observar que el resultado son 3 races, 2
imaginarias, y una real. Obviamente se descartaran cualquier valor imaginario
y cualquier valor negativo ya que el valor de z, est relacionado directamente
con el valor numrico del volumen.
z = u,7S2
El valor de z, entonces es:
El nico parmetro faltante para determinar el coeficiente de fugacidad en la
mezcla de la fase gaseosa es sigma, que lo calculamos con:
2 / 1
2
=
a
a
i
i
2
o
2
= 2,SS6
(f.3.22)
Solo hace falta calcular o debido que la constante de equilibrio que pide el
problema es la del benceno.
Con los valores determinados, sustituimos en la ecuacin (f.3.20) obteniendo
el coeficiente de fugacidad para el componente 2.
q
F
= . 74
t
|
L
) 1 (
,
) 0 (
, ,
log log log
L i i L i
o
L i
w + =
27055809 , 0 log
) 0 (
,
=
L i
25262407 , 0 log
) 1 (
,
=
L i
o
L i,
log = -u,S2S6u914
o
L i,
i
Clculo del coeficiente de fugacidad del lquido puro, q .
La ecuacin (4.40) nos permite resolver este parmetro.
(4.40)
=0,4747
. Clculo del coeficiente de Actividad,
Por la ecuacin (4.39) tenemos:
RT
v
C
i
j i iL
i
2
1
j
) (
ln
=
=
:
1
= 84,u
cm
3
mo
(4.39)
Se calculan los parmetros correspondientes. Los valores de los volumenes
molares y las solubilidades se leen directamente de la tabla, del anexo 4.1.
:
2
= 89,4
cm
3
mol l
o
1
= 6,4 _
col
cm
3
]
12
o
2
= 9,1S8_
col
cm
3
]
12
Se deben convertir las fracciones molares en fracciones lquidas.Para ello
debemos tener las densidades de los componenetes.
Tabla 4.4. Composicin lquida de los componentes.
Componente kgmol PM kg kg/m
3
m
3
xi
L
1 (n-C
3
) 0,109 44,097 4,81 507,199 9,48x10
-3
0,107
2 (C
6
) 0,891 78,114 69,6 883,917 7,874x10
-2
0,893
Fuente: Propia.
=
1
0,1012
=
2
0,89881
El coeficiente de actividad es:
007364 , 1
2
=
La constante de equilibrio es:
K
2
=0,6792
El valor reportado en la literatura K
2
=0,679; como podemos observar no hay
diferencia alguna en los clculos desarrollados en este problema.
Nomograma de DePriester
Uno de los mtodos ms sencillo de resolucin de problemas de
equilibrio de fases, es donde se determina la constante de equilibrio por el
Nomograma de DePriester. Esta correlacin, permite contar con efectos
promedios de composiciones, pero la base esencial es la ley de Raoult. La ley
de Raoult expresa los valores de K, como funciones simplemente de T y P,
independientes de las composiciones de las fases vapor y lquido. Cuando las
suposiciones que sirven de fundamento a la ley de Raoult son apropiadas,
permite que los valores de K se calculen y correlacionen como funciones de T y
P. Para mezclas de hidrocarburos ligeros y de otras molculas simples, en las
que los campos de fuerza moleculares no son complicados, las correlaciones de
esta clase tienen validez aproximada. La Fig. 4.7 y 4.8, muestra un
nomograma para valores de K de hidrocarburos ligeros como funciones de T y
P.
Fig. 4.7 Valores de K para los sistemas de hidrocarburos ligeros. Intervalo de
temperaturas baja. Fuente: Charts of C.L. DePriester. Chem. Eng. Prog. Symp
(1953).
Fig. 4.8 Cont. Valores de K para los sistemas de hidrocarburos ligeros.
Intervalo de temperaturas baja. Fuente: Charts of C.L. DePriester. Chem. Eng.
Prog. Symp (1953).
Condiciones de Puntos de Burbuja y Roco.
Para resolver ejercicios con el Nomograma de DePriester, es necesario que
recordemos las condiciones de punto de burbuja y roco, vistas en el tema 2.
La constante de equilibrio es:
K
=
y
= 1
n
= K
= K
1 = K
(4.41)
x
= 1
n
=
y
Punto de Burbuja.
La condicin de punto de burbuja es que la , ya que la composicin de
la fase lquida es dada, y la que se debe determinar es la de la fase de vapor.
Despejando yi de la constante de equilibrio:
Aplicando la condicin de burbujeo queda:
Punto de Roco.
La condicin de punto de roco es que la , ya que la composicin de la
fase vapor es dada, y la que se debe determinar es la de la fase de lquida.
Despejando xi de la constante de equilibrio:
1 =
y
Aplicando la condicin de roco:
(4.42)
PROBLEMA 5.
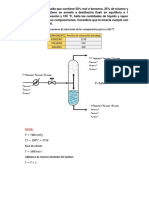
Un depsito de vaporizacin instantnea se alimenta con una mezcla
lquida de 42% molar de propano, y 58% molar de etano. En el
depsito se reduce la presin y se separan corrientes de L y V. El
sistema est a 305 K y ha de vaporizar 40% molar de lquido
suministrado. Cul es la presin a la que debe operar el depsito?
Cules son las composiciones en el equilibrio?
Solucin
T= 305K
P= ?
z
1
= 0,58
z
2
= 0,42
Asumiendo: B.C.= 100 kgmol de Alimentacin
V= 40 kgmol
Podemos establecer los lmites de la P, con el clculo de Presin de burbuja y
clculo de Presin de roco.
Presin de Burbuja: se aplica la condicin de la ecuacin (4.41). Asumiendo
la alimentacin como la composicin de la fase lquida nos queda:
z
1
= x
1
= 0,58
z
2
= x
2
= 0,42
Asumiremos valores de Presin de burbuja hasta que se cumpla la condicin de
(4.41).
Asumiendo P = 2800 kPa. Con la presin asumida y la T dada, se traza una
lnea entre la P y T en el nomograma, que corte cada una de las lneas de las
constantes del etano y propano. Las constantes de equilibrio se leen
directamente de la Fig. 4.7.
Tabla 4.5 P = 2800 kPa Condicin de Burbuja
Componente Ki zi Ki*zi
C2 1,5 0,58 0,87
C3 0,52 0,42 0,2184
Fuente: Propia
1,u884 = K
La condicin (4.41) no se cumple, se debe asumir un nuevo valor de P, que
disminuya la constante de equilibrio, para as disminuir la sumatoria.
Observando el nomograma, para que disminuya la constante de equilibrio debe
aumentar la presin. Asumiendo otro valor de P:
Tabla 4.6 3000kPa Condicin de Burbuja
Componente Ki zi Ki*zi
C2 1,38 0,58 0,8004
C3 0,49 0,42 0,2058
Fuente: Propia
1,uu62 = K
Como se cumple la condicin, la presin de burbuja es 3000 kPa.
Presin de Roco
Asumiendo las zi= yi, tenemos que:
z
1
= y
1
= 0,58
z
2
= y
2
= 0,42
Asumimos valor de P, hasta que la condicin (4.42) se cumpla.
Tabla 4.7 2500kPa Condicin de Roco
Componente Ki zi zi/Ki
C2 1,7 0,58 0,34
C3 0,58 0,42 0,72
Fuente: Propia
1,u6 =
y
La condicin (4.42) no se cumple, se asume un nuevo valor de P. Para
disminuir la sumatoria, se debe incrementar el valor de Ki, para que esto
ocurra, observemos la Fig. 4.7, se debe disminuir la P.
Asumiendo un nuevo valor de 2300 kPa se tiene:
Tabla 4.8 2300kPa Condicin de Roco
Componente Ki zi zi/Ki
C2 1,8 0,58 0,32
C3 0,62 0,42 0,68
Fuente: Propia
La presin de roco es 2300 kPa.
La presin del sistema, es la incgnita del problema, debe estar entre la
presin de burbuja y la de roco para que se vaporice el 40% de la
alimentacin.
Para determinar la P del sistema, se deben asumir valores de P, hasta que se
cumpla la ecuacin (2.53) de vaporizacin instantnea, estudiada en el tema
2.
1 =
z
1 +I (K
- 1)
Asumiendo P = 2700 kPa
Se leen las constantes de equilibrio en el nomograma Fig. 4.7, a la presin de
2700 kPa y la temperatura del sistema 31,85 C.
Tabla 4.9 2700kPa Vaporizacin
Componente Ki zi yi
C2 1,51 0,58 0,727
C3 0,52 0,42 0,270
Fuente: Propia
Ya que:
1 + I (K
-1)
= u,998
La presin del sistema es 2700 kPa.
4.7. DETERMINACIN DE PUNTOS DE BURBUJA Y ROCO. SEPARACIN
INSTANTANEA, COMPORTAMIENTO REAL.
Lo expuesto anteriormente en el tema 2, se ha restringido a sistemas ideales
donde las constantes de equilibrio slo dependen de la temperatura, y se
calculan mediante las presiones de vapor de los componentes. Sin embargo la
adopcin de un modelo de equilibrio de fases no-lineal trae la complicacin
aadida de que las relaciones de equilibrio dependen tambin de la
composicin. El modelo de flash generalizado, viene dado en ese caso por las
ecuaciones mostradas en la tabla 4.10.
Tabla 4.10. Ecuaciones relacionadas a
problemas de equilibrio de fases.
Ecuaciones Descripcin
z
F = I y
+I x
(a) balance por componentes
F = I + I (b) balance de masa global
y
= K
(c) constante de equilibrio
K
=
y
(x, I)
0
(I, P)
v
(y, I) P
(u)
- y
constante de equilibrio mtodo
y
- x
= u (e) Condicin
FE
]
+ = IE
(y, I, P) + IE
I
(x, I, P) (f) Balances de energa
Fuente: opia
Dependiendo de las especificaciones del problema, el proceso de clculo tendr
mayor o menor complejidad, tal y como tambin sucede en el modelo ideal.
Pero dadas las no-linealidades inherentes al modelo, es necesario seguir un
procedimiento iterativo, por lo que se recomienda la utilizacin de la
Pr
programacin por ordenador. (J. Snchez).
Tambin ser necesario en este caso disponer de un acceso a banco de datos
termodinmicos (fugacidades, coeficientes de actividad, etc.).
tiene un primer
clculo simplificado del flash, est especialmente recomendada.
s se presentan
a continuacin, basados en la ecuacin de equilibrio para sistemas ajustados al
K
=
y
i
P
La simulacin en Aspen, Pro-II, entre otros, una vez que se
Los diagramas de flujo para los problemas de equilibrio de fase
mtodo - y:
(4.29)
Sustituyendo la ecuacin (3.38a) del tema 3, en la ecuacin (4.29) se tiene:
que:
sut
P y
= x
sut
( 4.4S)
Ya
= cxp _
-:
L
(P - P
sut
)
RI
_ (4.44)
sin dentro del corchete, es el factor de correccin de Poyting, a bajas y
moderadas presiones tiende a la unidad, entonces la ecuacin (4.44) se expresa
como:
La expre
sut
(4.4S)
La constante de equilibrio proveniente de la ecuacin (4.43), conviene
representarla entonces como (4.46) para clculos computacionales:
K
=
y
sut
P
(4.46)
lquida. (T,
x
1
, x
2
, x
3
, .. x
n
).
a Fig. 4.9 representa el algoritmo computacional para la resolucin de la
osiciones de vapor y la P.
Presin de Burbuja: Dada Temperatura y composicin de la fase
L
presin de burbuja. Dada la T, y la composicin de la fase lquida, se
determinan las comp
Fig.4.9 Diagrama de Flujo para el clculo de Presin de Burbuja.
Fuente: Smith. Sptima edicin
El programa comienza leyendo los datos de T, composicin de la fase lquida,
constantes de Antoine.
1. Para la primera iteracin los
se asumen como 1.
2. S
s, a la T y composicin de la fase lquida dada.
) proveniente de la
1
e calculan las presiones de saturacin por Antoine a la T dada.
3. Se calculan los coeficientes de actividad por algunos de los modelos de
solucin estudiado
4. Se calcula la P del sistema por la ecuacin (4.49
ecuacin de equilibrio (4.43):
P y
= x
sut
( 4.4S)
P y
1
=
y
1
P
1
sut
x
1
(4.47)
P y
2
=
y
2
P
2
sut
x
2
2
(4.48)
umando (4.47) y (4.48), y generalizando para sistemas multicomponentes se
tiene:
P =
y
sut
x
S
(4.49)
las fracciones de vapor calculadas, se calculan los
con (4.44)
(4.45) segn sea el caso.
7. Se calcula nuevamente la P con la ecuacin (4.49). Esta vez tomando en
cuenta los
calculados.
s decir el cambio de la P,
de una interaccin a otra, sea menor a la tolerancia (e).
5. Se calculan las fracciones de vapor con (4.43).
6. Con
8. La iteracin contina hasta que el delta de la P, e
Presin de Roco: Dada Temperatura y composicin de la fase vapor. (T, y
1
,
omposicin de la
de vapor.
r calculados porque estos dependen de la composicin de la
fase lquida).
(4.43).
x
1
=
1 1
y
1
P
1
sut
y
2
, y
3
, .. y
n
).
El algoritmo de la Fig. 4.10, comienza leyendo los datos necesarios para este
clculo, los cuales son la T, las constantes de Antoine, y la c
fase
1. Se fijan
=1, del mismo modo los coeficientes de actividad (ya que no
pueden se
2. Se calculan las presiones de saturacin por Antoine.
3. Se calculan la P por la ecuacin (4.52) proveniente de la ecuacin de
equilibrio
P y
= x
sut
( 4.4S)
P y
(4.Su)
P
x
2
=
2 2
y
2
P
2
sut
y
(4.S1)
Sumando (4.50) y (4.51), y rearreglando para ecuacin general para
multicomponentes:
P =
1
y P
sut
(4.S2)
Fig. 4.10 Diagrama de Flujo para el clculo de Presin de Roco.
Fuente: Smith. Sptima edicin
4. Con la P calculada, se calculan las composiciones de la fase lquida con
(4.43).
5. Con las fracciones lquidas determinadas se calculan los y
por el modelo
de solucin seleccionado.
6. Se calcula nuevamente la P con la ecuacin (4.52) con los y
calculados.
7. Se determinan los
.
8. Se calculan las fracciones de la fase lquida con (4.43). Si los valores de
x
xi, normalizados se calculan nuevamente los
coeficientes de actividad con el modelo de solucin seleccionado.
el calculado en 9) a la tolerancia (e)?. Si es no, nos devolvemos
.
i, se termina.
rabajara
os derechos de la
ica por la presin de saturacin (fuera de la
P
]
sut
=
P
y
]
x
]
i, no suman 1, se normalizan.
9. Con los valores de
10. Evaluamos el delta de los y
. Es menor el oy
(se resta el y
calculado en
5, menos
a 8, con los y
calculados en 9. Si es si, se sigue a 11.
11. Se calcula la P, con (4.52)
12. Evaluamos el delta de P. es menor el oP a la tolerancia?. Si es no,
regresamos a 7. Si es s
Cuando el sistema, no especfica la T, los clculos suelen ser ms trabajosos,
ya que se prestan a que sean tratamientos iterativos, por ello se t
tanto para la temperatura de burbuja como la temperatura roco con
relaciones de presiones de vapor ya que son funciones dbiles de la
temperatura. Para introducir estas razones en los miembr
ecuacin (4.49) y (4.52), se multipl
sumatoria) y se divide (dentro de la sumatoria).
_
P
sut
P
sut
]
_
n
]
(4.SS)
P
]
sut
= P
y
]
y
]
_
P
]
sut
P
sut
_
n
]
(4.S4)
j: es un componente seleccionado arbitrariamente.
La tolerancia ser:
Iolor (n) - :olor (n - 1) < e
e = u,u1 :olor( n)
Temperatura de Burbuja: Dada la Presin y composicin de la fase lquida.
(P, x
1
, x
2
, x
3
, .. x
n
).
La Fig. 4.11 muestra el algoritmo utilizado para la programacin con
herramientas computacionales.
culo, los cuales son la
Antoine, y la composicin de la fase lquida.
1. Se fijan
=1.
2. Se calculan las Temperaturas de satur cin por Antoine a la P del
era T por (4.55), para comenzar la iteracin.
I = x I
sut
(4.SS)
de saturacin por Antoine a la Temperatura
asumida en 3.
especie j, para calcular la P
j
por la ecuacin (4.53).
calculada en 7, se determinan las presiones de saturacin por
9. Se calculan los y
i
de la ecuacin (4.43).
10. Con los yi calculado 9, se determina los
.
on los y
calculados en 11, con yi calculados en 9, y los
Se comienza leyendo los datos necesarios para este cl
P, las constantes de
a
sistema.
3. Se calcula una prim
4. Se calculan las presiones
5. Se calculan los y
por el modelo de solucin seleccionado, a la T
asumida, y composicin de la fase lquida dada.
6. Se selecciona la
7. Se calcula la T, con la ecuacin de Antoine a la P
j
, calculada en 6.
8. Con la T
Antoine.
s en
11. Se calculan los y
a la T calculada en 7.
12. Se calcula Pj, c
calculados en 10.
13. Se calcula la T mediante la ecuacin de Antoine a la Pj calculada en 12.
a otro
lo de Temperatura de Burbuja.
ima edicin
Temperatura de Roco: Dada la Presin y composicin de la fase de vapor.
(P, y
1
, y
2
, y
3
, .. y
n
).
Fig. 4.12 Diagrama de Flujo para el clculo de Temperatura de Roco.
Fuente: Smith. Sptima edicin
La Fig. 4.12 muestra el algoritmo utilizado para la programacin con
herramientas computacionales.
Se comienza leyendo los datos necesarios para este clculo, los cuales son la
P, las constantes de Antoine, y la composicin de la fase de vapor.
Es la el delta de T menor a la tolerancia (e)? Si es no, se tom
valor de T desde 7. Si es si, termina.
Fig.4.11 Diagrama de Flujo para el clcu
Fuente: Smith. Spt
1. Se fijan
=1 y los y
= 1
2. Se calculan las Temperaturas de saturacin por Antoine a la P del
sistema.
3. Se calcula una primera T por (4.56), para comenzar la iteracin.
I = y
sut
(4.S6)
4. Se calculan las presiones de saturacin por Antoine a la Temperatura
asumida en 3.
5. Se selecciona la especie j, para calcular la P
j
por la ecuacin (4.54).
6. Se calcula la T, con la ecuacin de Antoine a la P , calculada en 5.
7. Con la T calculada iones de saturacin por
Antoine.
acin (4.43).
10. Se calculan los y
por el modelo de solucin seleccionado, a la T
lados en 9,
Pi
sat
y
, a la T calculada en 12.
lados en
13, los y
determinados en 10. Si la sumatoria no da 1, se normalizan
los valores de xi.
15. Se calculan los valores de y
, con xi calculados en 14 y la T calculada en
12.
16. Es cada delta de y menor a la tolerancia. Si es no, nos devolvemos a
14, con los nuevos valores de y calculados en 15. Este paso se repite
hasta que se cumpla la tolerancia. Si es si, seguimos a 17.
17. Se calcula Pj (4.54), con los y
calculados en 16 que cumplan con la
tolerancia, con xi calculados en 16, y los
calculados en 8.
j
en 6, se determinan las pres
8. Se calculan los
.
9. Se calculan los x
i
de la ecu
asumida en 6 y los xi calculados en 9.
11. Se calcula Pj (4.54), con los y
calculados en 10, con xi calcu
y los
calculados en 8.
12. Se calcula la T mediante la ecuacin de Antoine a la Pj calculada en 11.
13. Se evala
14. Se calculan xi por (4.43) tomando los valores de
y Pi
sat
calcu
18. Se calcula la T, con Antoine a la Pj calculada en 17. Es el delta de T
menor a la tolerancia? Si es no, tomamos otro valor de T desde 6. Si es
esolver los clculos de vaporizacin instantnea, es la
lobales, y balances por componentes realizados al sistema, e
si, se termina.
Vaporizacin Instantnea.
La ecuacin para r
misma que hemos estudiado desde el tema 2 la ecuacin (2.52), proveniente
de balances g
introduciendo el concepto de la constante de equilibrio.
y
=
z K
1 + I (K
- 1)
(2.S2)
Aplicando sumatoria a ambos lados de la ecuacin:
1 =
z
1 + I (K
-1)
(2.SS)
Como la constante de equilibrio, para sistemas reales, depende
ertemente de las composiciones de las fases en coexistencia, no puede ser
ibrio de Raoult, al menos que las condiciones lo
ermitan. Las constantes de equilibrio pueden estar determinadas por
cualqu
e realiza el balance en
ncin de la composicin de la fase lquida quedando:
x
=
K
1 +I (K
- 1)
fu
utilizada la constante de equil
p
iera de las ecuaciones estudiadas en el apartado 4.6 de este mismo
tema.
Una ecuacin anloga a la ecuacin (2.52) es cuando s
fu
(4.S7)
la 4.10, Rachford y Rice
determinan una ecuacin ms conveniente para los clculos.
Aplicando la ecuacin (e) que aparece en la tab
Rachford y Rice
Estos autores desarrollaron otra forma de la ecuacin como funcin
objetivo para la convergencia:
y
i
- x
i
= 0 x
i
- y
i
= 0
Donde V de (4.57) y (XXX) se convierte en una relacin del los moles de vapor
con la alimentacin es decir V/F.
=
+
+
=
i i i
i
i
i i
F V K
z
F V K
z K
F V f 0
/ ) 1 ( 1 / ) 1 ( 1
) / (
=
+
=
i i
F V K
K z
F V f 0
/ ) 1 ( 1
) 1 (
) / ( (4.58)
i i
La funcin f(V/F) tiene las siguientes caractersticas:
- No tiene ni mximos ni mnimos, ni races falsas
- f(V/F) es aproximadamente lineal
La condicin de fase se puede establecer a partir de la ecuacin que aparecen
el la tabla 4.12 f(V/F):
f(1) Condicin de fase
Tabla 4.12. Condicin de fase.
f(0)
< ado 0 <0 Lquido sub-enfri
=0 <0 Lquido saturado
>0 <0 Mezcla vapor-lquido
>0 =0 Vapor saturado
>0 >0 Vapor sobrecalentado
Fuente: Snchez J.
ergencia descritos en el
cin) para hallar la solucin;
Newton-Raphson:
Se pueden utilizar los procedimientos de conv
Apndice I, del libro gua (Smith y Col, sptima edi
por ejemplo si se utiliza el mtodo de
[ ]
1
2
2
'
'
1
) / (
/ ) 1 (K
i
1
) 1
) / (
) / (
(
) / ( ) / (
<
+
=
=
+
F V f
F V
F V f
F V
V f
F V F V
i
i
k
k
(4.59)
(K z
i
) / F
k
f
k
2
1
) / (
) / ( ) / (
<
+
k
k k
F V
F V F V
13 describe el algoritmo computacional utilizado para este tipo de
lculos.
Fig. 4.13 Diagrama de Flujo para el clculo de P, T de vaporizacin
instantnea.
Fuente: Smith. Sptima edicin
La fig. 4.
c
Dada la T y P, determinar las composiciones de las fases en el equilibrio.
1. Se calcula la presin de burbuja y roco, por las Fig. 4.10 y 4.11.
2. Con estos clculos se estiman valores aproximados para y
.
3. Con los valores inciales, se calculan Ki a travs de la ecuacin (4.46).
4. Se calculan (V/F) por la ecuacin (4.58). Si no converge se aplica el
mtodo de Newton Raphson.
5. El nuevo valor para la iteracin ser donde (4.59) es su derivada:
k
k
k k
F V f
F V f
F V F V
) / (
) / (
) / ( ) / (
' 1
=
+
6. Con el valor de V/F determinado se calculas las xi y las yi. Cada uno de
los valores de V/F, xi y yi, deben ser menor a la tolerancia. Cuando se
cumpla se termina la iteracin.
PROBLEMA 6.
Determine para el sistema 1-Propanol(1)/ Agua (2) la Temperatura de
burbuja, P=110 kPa, x1= 0,25.
Aplique Wilson para la fase lquida y para la fase de vapor la ecuacin virial por
itzer Curl. Los datos suministrados son:
Compon
21
P
Tabla 4.13 Parmetros de Wilson
m3/mol) o entes v(c
12
o
1
775,48 1351,9
75,14
2 18,07
Fuente: Smith y Col.
Las propiedades crticas son las expresadas en la tabla 4.14.
Tabla 4.14 Propiedades Crticas para el sistema 1 Propanol (1)/Agua (2)
Componentes Tc K Pc kPa zc w Vc m3/kgmol
1 536,8 5175 0,219 0,254 0,622
2 647,1 22055 0,0559 0,229 0,345
Fuente: Smith y Col.
Las contantes de Antoine se observan en la tabla 4.15:
Tabla 4.15 Constantes de Antoine para el sistema 1 Propanol (1)/Agua (2)
Componentes
A B C
1 15,5289 3166,38 80,15
2 16,2886 3816,44 46,13
Fuente: Gama M.
el flujograma mostrado en Fig. 4.11. Para
llo se utilizo la hoja de clculo de Excel, facilitando de este modo, los clculos.
licado.
Tabla 4.16. edimiento empleado en fig. 4.11
Se realizo el procedimiento explicado para determinar Temperatura de
Burbuja, siguiendo lo explicado en
e
A continuacin se muestra la hoja de clculo (tabla 4.16), en la cual cada celda
represento un paso del procedimiento exp
Resultados d oc el pr
1 1
2 1
T
1
sat
372,5638616
T
2
375,470749
sat
Tcalculada(4.55)
374,7440272
P
1
sut
119,1778496
P
2
sut
107,2168612
12 0,084886166
21 0,676865235
lny
1 0,810138558
lny
2 0,200664604
y
1 2,248219473
y
2 1,222214777
(2) 71,36285557
T(n1)
363,6157628
P
1
sut
78,15250623
P
2
s
71,36285557
ut
y1
0,399327242
y2
0,59468684
1 0,989641354
2 0,995114236
12 0,082223518
21 0,640284989
lny
1 0,836015289
lny
2 0,2041666
y
1 2,307155288
y
2 1,226502471
(2) 70,39229856
T(n)
363,2545072
El delta
T(n)- T(n-1) < e
Se calcula el e = u.u1 S6S,2S4Su72
,632545
,63
Como se cumple, la temperatura de burbuj a T
B
=363,2545072 K
T
B
= 90,1045
Otra herramienta rpida y precisa es el simulador PRO-II, donde comprobamos
este resultado. En las Fig 4.14-4.15 podem r algunas de las pantallas
del programa. Se selecciona los compo la mezcla y el mtodo
termodinmico. Haciendo clip en la corriente de alimentacin del separador, el
programa arroja una ventana cual se ron los datos de P y ,
como segunda especificacin se coloco Bu burbuja). En la
pestaa que dice Flowarate an compos o y composicin), se le
suministraron los datos de comp cin de f .
4. Simulador PRO-II. Seleccin de componentes y mtodo
termodinmic lla PRO-II
de T, ser:
e =3 072
0,36125561<3 2545072
a, es igual
07 C
os observa
nentes de
en el le suministra
bble Point (punto de
d itions (fluj
osi ase lquida
Fig.4.1
o. Fuente: Panta
Fig. 4.15. Simulador de procesos PRO-II. Calculo de Temperatura de Burbuja.
Fuente: Propia.
Los resultados arrojados por el programa los podemos observar en la Fig.
4.16.
La temperatura determinada por el simulador es de: 90,21 C. La poca
diferencia se debe, a que el mtodo termodinmico seleccionado en el
simulador fue Wilson para la fase lquida e idealidad para la fase gaseosa, es
decir que los coeficientes de fugacidades el simulador los tomo como 1. Sin
embargo en el algoritmo planteado, se calcularon cada uno de los
por la ecuacin de Pitzer. Como hemos visto la presin del sistema es baja
110 kPa (1,10 bar), lo cual nos permite concluir que el clculo de coeficientes de
fugacidad pueden ser obviados.
REFERENCIAS
1. Balzhiser R. Michael R.S. Termodinmica Qumica para Ingenieros.
Editorial Pretince-Hall Internacional. 1974
2. Crotti, M. Por qu se produce la condensacin retrograda. Temas de
geniera de Reservorios. |Documento en lnea Disponible:
http://www.inlab.com.ar/Cond_retr.htm| Consulta 2006, Marzo, 15|
3. PhD. Yolanda Reyes. Princ
In
ipios De Termodinmica Aplicada Para El rea
e Ingeniera Qumica.
4. Praunitz J.M. Molecular Thermodynamics of Fluids Phase Equilibria.
Chap. 5, Pretince-Hall. 1969.
5. Smith Van Nees. Introduccin a la Termodinmica en Ingeniera Qumica.
6ta Edicin. 1996. Editorial McGraw Hill
6. Termodinmica Ingeniera Qum a. Profesora titular PhD. Luz Marina
Jaramillo. Santiago de Cali, 2001.
D
.
ic
S-ar putea să vă placă și
- Teoría Del Gas Real: La Relación Exacta Para Gases RealesDe la EverandTeoría Del Gas Real: La Relación Exacta Para Gases RealesÎncă nu există evaluări
- Tema IVDocument56 paginiTema IVMichaell LinaresÎncă nu există evaluări
- Problema 7.1Document8 paginiProblema 7.1Lalommoreno Moreno OvandoÎncă nu există evaluări
- Punto EutecticoDocument9 paginiPunto EutecticoYunuen TapiaÎncă nu există evaluări
- Talller Dest y Secado 2019ADocument6 paginiTalller Dest y Secado 2019AFrancisco OrozcoÎncă nu există evaluări
- Preinforme Punto de Burbuja 1Document7 paginiPreinforme Punto de Burbuja 1Magaly PinzónÎncă nu există evaluări
- Práctica 4 Dilatometria Laboratorio Integral IiDocument13 paginiPráctica 4 Dilatometria Laboratorio Integral IiRodolfo Luis LimónÎncă nu există evaluări
- Practica Num 4Document5 paginiPractica Num 4victacito100% (1)
- Indice de Refraccion Esta en InternetDocument13 paginiIndice de Refraccion Esta en InternetLuis Cesar Gutierrez MedranoÎncă nu există evaluări
- LixiviaciónDocument8 paginiLixiviaciónSaudy MezaÎncă nu există evaluări
- 03 Problemas-1Document3 pagini03 Problemas-1María de la Luz VeraÎncă nu există evaluări
- Absorcion No IsotermicaDocument6 paginiAbsorcion No IsotermicaNatalia Moreno MorenoÎncă nu există evaluări
- Introducción A La Destilación Con ReflujoDocument3 paginiIntroducción A La Destilación Con ReflujoFuerez A-yÎncă nu există evaluări
- Diagrama de Fases de Un Sistema TernarioDocument13 paginiDiagrama de Fases de Un Sistema TernarioJohn Montellanos100% (2)
- Metodo para La Destilación Batch Binario Considerando Una Politica de Reflujo VariableDocument5 paginiMetodo para La Destilación Batch Binario Considerando Una Politica de Reflujo VariableWilmerRamirezSalvadorÎncă nu există evaluări
- Laboratorio N°4Document21 paginiLaboratorio N°4FRANSÎncă nu există evaluări
- Columnas de Rectificacion 23Document25 paginiColumnas de Rectificacion 23Rodrigo Salinas HuamancondorÎncă nu există evaluări
- Tarea 2Document6 paginiTarea 2Ricky Ocampo0% (1)
- Propiedades Molares ParcialesDocument17 paginiPropiedades Molares ParcialesDaisy ChannelsÎncă nu există evaluări
- Informe 10Document24 paginiInforme 10nubrolitoÎncă nu există evaluări
- Practica 6Document19 paginiPractica 6Ray AcostaÎncă nu există evaluări
- CumenoDocument6 paginiCumenoJoaquin Valdez FernandezÎncă nu există evaluări
- ACETONA AGUA DestilacionDocument16 paginiACETONA AGUA DestilacionBryan Carrera0% (1)
- Equilibrio Solido PracticaDocument10 paginiEquilibrio Solido PracticaFrancisco Madrigal Meraz100% (1)
- Destilación Método Doble Efecto-GuiaDocument23 paginiDestilación Método Doble Efecto-GuiaLinaSofiaÎncă nu există evaluări
- PERVAPORACIONDocument27 paginiPERVAPORACIONLuis100% (1)
- Practica 1Document17 paginiPractica 1Rodrigo100% (1)
- Deshidratación Catalizada de D-Xilosa para La Obtención de FurfuralDocument4 paginiDeshidratación Catalizada de D-Xilosa para La Obtención de FurfuralFernando HuarocÎncă nu există evaluări
- Columna de Rectificacion de Platos Con Cachucas de BurbujeoDocument16 paginiColumna de Rectificacion de Platos Con Cachucas de BurbujeoMartin Nicolas Trinidad GonzalezÎncă nu există evaluări
- Informe Descomposición Del Peróxido de HidrógenoDocument4 paginiInforme Descomposición Del Peróxido de HidrógenoISABELA SOLARTE CASTILLOÎncă nu există evaluări
- LAB OP1. Practica 6Document29 paginiLAB OP1. Practica 6Jesus David OcandoÎncă nu există evaluări
- Problemas Resueltos de MetalurgiaDocument5 paginiProblemas Resueltos de MetalurgiaJoseÎncă nu există evaluări
- 04 Introduccion Al Uso de Simulador Coco I-2021Document14 pagini04 Introduccion Al Uso de Simulador Coco I-2021Eugenia Zurita GalarzaÎncă nu există evaluări
- Separador Flash Con Recirculación PDFDocument11 paginiSeparador Flash Con Recirculación PDFamerico007Încă nu există evaluări
- Balance de Masa en Sistemas ReactivosDocument4 paginiBalance de Masa en Sistemas ReactivosRaul ErnestoÎncă nu există evaluări
- Reactor So3Document50 paginiReactor So3williamyc7Încă nu există evaluări
- Microsoft PowerPoint - Problemas Resuelt PDFDocument6 paginiMicrosoft PowerPoint - Problemas Resuelt PDFMarckos Ramos AbadÎncă nu există evaluări
- Destilacion UnitariasiiDocument25 paginiDestilacion UnitariasiiganimedesÎncă nu există evaluări
- Métodos RigurososDocument4 paginiMétodos Rigurosossilver---ioriÎncă nu există evaluări
- Actividad DE FISICOQUIMICADocument6 paginiActividad DE FISICOQUIMICAAvelin CastillaÎncă nu există evaluări
- Practica 1Document45 paginiPractica 1Rocio FloresÎncă nu există evaluări
- 5.titulaciones ConductimetricasDocument12 pagini5.titulaciones Conductimetricasjofre232450% (2)
- Apuntes Temas 2 9 FisicoquimicaDocument127 paginiApuntes Temas 2 9 FisicoquimicaVilchis Barajas Uriel100% (1)
- Regla 3 ComponentesDocument2 paginiRegla 3 ComponentesPamela Sanhueza JaraÎncă nu există evaluări
- Investigacion Tipos de Valvulas.Document6 paginiInvestigacion Tipos de Valvulas.Oscar Gia BeldumaÎncă nu există evaluări
- Transferencia de MasaDocument5 paginiTransferencia de MasaELIZABETHÎncă nu există evaluări
- Taller DestilaciónDocument3 paginiTaller DestilaciónDaniela MontenegroÎncă nu există evaluări
- Edoc - Pub - Combustion Ucsm 2010 CFB PDFDocument110 paginiEdoc - Pub - Combustion Ucsm 2010 CFB PDFANTHONY PERCY INGA HUANCAÎncă nu există evaluări
- Manual Herramienta - Reactor BatchDocument12 paginiManual Herramienta - Reactor BatchSebastian GaitanÎncă nu există evaluări
- Construcción de La Tabla Estequiometria para Sistemas VariablesDocument3 paginiConstrucción de La Tabla Estequiometria para Sistemas VariablesEdgar Gabriel OrtizÎncă nu există evaluări
- Practica 8 Laboratorio Integral IIDocument8 paginiPractica 8 Laboratorio Integral IIErick HernándezÎncă nu există evaluări
- Producción de Anhídrido MaleicoDocument4 paginiProducción de Anhídrido MaleicoJosu VillAlobosÎncă nu există evaluări
- PS 1-2008 2do ParcialDocument2 paginiPS 1-2008 2do ParcialRosmaryan GuzmanÎncă nu există evaluări
- Expo de Coca 6Document6 paginiExpo de Coca 6KevinEricksonSilvaIsidroÎncă nu există evaluări
- Problema 3. AbsorciónDocument15 paginiProblema 3. AbsorciónCarlosm Mata DiazÎncă nu există evaluări
- Tema - IV (Ejercicio L-V) 3Document57 paginiTema - IV (Ejercicio L-V) 3Jenny Gema Citizen VilesÎncă nu există evaluări
- DePriester (K de Equilibrio 2)Document61 paginiDePriester (K de Equilibrio 2)Maria Quintero VillasmilÎncă nu există evaluări
- Regla de FasesDocument7 paginiRegla de Fasesrompecaminos02Încă nu există evaluări
- Diagrama Liquido VaporDocument57 paginiDiagrama Liquido VaporshlainnÎncă nu există evaluări
- Equilibrio de Mezaclas Multi ComponentesDocument31 paginiEquilibrio de Mezaclas Multi ComponentesMario Ramiro Humerez Diaz100% (2)
- Contreras (2020) - Las Líneas de Investigación Desde La Perspectiva de Funcionalidad para Los Estudiantes de Postgrado de La Universidad VenezolanaDocument12 paginiContreras (2020) - Las Líneas de Investigación Desde La Perspectiva de Funcionalidad para Los Estudiantes de Postgrado de La Universidad VenezolanadanifexÎncă nu există evaluări
- Tarifas Y Comisiones Por Servicio Vigente A Partir Del 14 de Noviembre de 2022Document9 paginiTarifas Y Comisiones Por Servicio Vigente A Partir Del 14 de Noviembre de 2022danifexÎncă nu există evaluări
- SCHA1301 - 033251-1-65 EsDocument85 paginiSCHA1301 - 033251-1-65 EsdanifexÎncă nu există evaluări
- Actividad 1. Daniel FalcónDocument4 paginiActividad 1. Daniel FalcóndanifexÎncă nu există evaluări
- Medidores (Instrumentacion)Document16 paginiMedidores (Instrumentacion)danifexÎncă nu există evaluări