S-ar putea să vă placă și
- DentistDocument1 paginăDentistLaura KristyÎncă nu există evaluări
- Doctor Patient RelationshipDocument21 paginiDoctor Patient RelationshipLaura KristyÎncă nu există evaluări
- Opioid Analgesics and AntagonistsDocument18 paginiOpioid Analgesics and AntagonistsLaura KristyÎncă nu există evaluări
- Atraumatic/ Alternative Restorative TreatmentDocument19 paginiAtraumatic/ Alternative Restorative TreatmentLaura KristyÎncă nu există evaluări
- Jurnal Lmx-KepuasanDocument18 paginiJurnal Lmx-KepuasanLaura KristyÎncă nu există evaluări
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5795)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (74)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- Consumer Information On Proper Use of YogaDocument168 paginiConsumer Information On Proper Use of Yogaskwycb04Încă nu există evaluări
- Antibullying Presentation 1Document23 paginiAntibullying Presentation 1Martin Ceazar HermocillaÎncă nu există evaluări
- Ratio & Proportion Part 1Document5 paginiRatio & Proportion Part 1P Singh KarkiÎncă nu există evaluări
- Govt Considers Putting ShahbazDocument27 paginiGovt Considers Putting ShahbazWanderer123Încă nu există evaluări
- HCT Baniqued P.D.E. Paper1 Version3 FullpaperDocument8 paginiHCT Baniqued P.D.E. Paper1 Version3 FullpaperJoshua HernandezÎncă nu există evaluări
- Effectives of e Wallets NewDocument15 paginiEffectives of e Wallets NewRicardo SantosÎncă nu există evaluări
- Of Gods, Glyphs and KingsDocument24 paginiOf Gods, Glyphs and KingsBraulioÎncă nu există evaluări
- Program 2019 MTAPTL Annual Convention PDFDocument3 paginiProgram 2019 MTAPTL Annual Convention PDFrichardlfigueroaÎncă nu există evaluări
- The Lion and The Boar Story EnglishDocument2 paginiThe Lion and The Boar Story EnglishKemal AmarullahÎncă nu există evaluări
- Earthquake Lesson Plan 2022Document5 paginiEarthquake Lesson Plan 2022Maylyn Grace Dalumpines-Colon EbonaloÎncă nu există evaluări
- This I BeleiveDocument3 paginiThis I Beleiveapi-708902979Încă nu există evaluări
- Peter Zumthor - Thinking Architecture PDFDocument33 paginiPeter Zumthor - Thinking Architecture PDFDiana Sterian73% (11)
- Distortion of The Ecclesiological Views of Metropolitan Chrysostomos of PhlorinaDocument11 paginiDistortion of The Ecclesiological Views of Metropolitan Chrysostomos of PhlorinaHibernoSlavÎncă nu există evaluări
- Reflective Journal 4Document3 paginiReflective Journal 4api-550030025Încă nu există evaluări
- Professional Education: St. Louis Review Center, IncDocument10 paginiProfessional Education: St. Louis Review Center, IncEarshad Shinichi IIIÎncă nu există evaluări
- Glide Reflection Folding InstructionsDocument1 paginăGlide Reflection Folding Instructionsapi-355107616Încă nu există evaluări
- Global Perspectives Reflective PaperDocument3 paginiGlobal Perspectives Reflective PaperMoaiz AttiqÎncă nu există evaluări
- VIETTELDocument20 paginiVIETTELSolgrynÎncă nu există evaluări
- Answers & Solutions: For For For For For JEE (MAIN) - 2019 (Online) Phase-2Document22 paginiAnswers & Solutions: For For For For For JEE (MAIN) - 2019 (Online) Phase-2Manila NandaÎncă nu există evaluări
- Settlement Geography: Unit No-1&2Document11 paginiSettlement Geography: Unit No-1&2Arindam RoulÎncă nu există evaluări
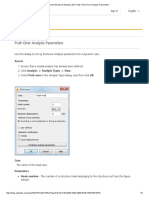
- Robot Structural Analysis 2017 Help - Push Over Analysis ParametersDocument3 paginiRobot Structural Analysis 2017 Help - Push Over Analysis ParametersJustin MusopoleÎncă nu există evaluări
- SwahiliDocument7 paginiSwahiliMohammedÎncă nu există evaluări
- J2 P IntroductionDocument9 paginiJ2 P IntroductionnitinvnjÎncă nu există evaluări
- University of Physician V CIRDocument24 paginiUniversity of Physician V CIROlivia JaneÎncă nu există evaluări
- EXERCISE 1-Passive FormDocument5 paginiEXERCISE 1-Passive FormMichele LangÎncă nu există evaluări
- Once Upon A Timein AmericaDocument335 paginiOnce Upon A Timein Americaqwerty-keysÎncă nu există evaluări
- BoSY CRLA Grade 1 MT Administration GuideDocument13 paginiBoSY CRLA Grade 1 MT Administration GuideJOCELYN SANANO100% (1)
- Math Studies Financial MathsDocument7 paginiMath Studies Financial MathsGirish MishraÎncă nu există evaluări
- Clause: Extra Element + Independent Clause Dependent ClauseDocument1 paginăClause: Extra Element + Independent Clause Dependent ClauseTieng HuangÎncă nu există evaluări
- IFRS Session 1 To 3Document40 paginiIFRS Session 1 To 3techna8Încă nu există evaluări