S-ar putea să vă placă și
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
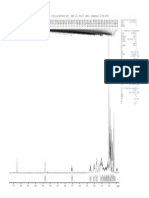
- F10 11 P51V (Otilia/gustavo Ufc) (RMN 1H, CDCL3) Oper.: Edangelo 21/08/2008Document1 paginăF10 11 P51V (Otilia/gustavo Ufc) (RMN 1H, CDCL3) Oper.: Edangelo 21/08/2008Gustavo Lima AlmeidaÎncă nu există evaluări
- Logk1 Predictor ManualDocument3 paginiLogk1 Predictor ManualGustavo Lima AlmeidaÎncă nu există evaluări
- Retention Time: PDA-475nm Licopeno t5r2 14Document2 paginiRetention Time: PDA-475nm Licopeno t5r2 14Gustavo Lima AlmeidaÎncă nu există evaluări
- 8PV33P2 6DDocument1 pagină8PV33P2 6DGustavo Lima AlmeidaÎncă nu există evaluări
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5795)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (74)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1091)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- SA164YKDocument6 paginiSA164YKperoz_ak47Încă nu există evaluări
- Learner Analysis Data DiscussionDocument5 paginiLearner Analysis Data Discussionapi-345451244Încă nu există evaluări
- Các chỉ số ảnh hưởng chỉ số môi trường đầu tưDocument13 paginiCác chỉ số ảnh hưởng chỉ số môi trường đầu tưK59 Nguyen Dang VuÎncă nu există evaluări
- Plastic To The Basket! A Community Project - 1Document25 paginiPlastic To The Basket! A Community Project - 1Rojas, Jasmine Rose LÎncă nu există evaluări
- CHRO Lab PDFDocument10 paginiCHRO Lab PDFChristian Cherry TeaÎncă nu există evaluări
- Models and Methods in Social Network Analysis: February 2005Document3 paginiModels and Methods in Social Network Analysis: February 2005Fajrin Nur HidayatÎncă nu există evaluări
- Market Research ProjectDocument27 paginiMarket Research ProjectAkhil AroraÎncă nu există evaluări
- Stat Q3 WK4 Las3Document1 paginăStat Q3 WK4 Las3Gladzangel LoricabvÎncă nu există evaluări
- 2021 Whistleblowing Benchmark Report Web Final1-1Document23 pagini2021 Whistleblowing Benchmark Report Web Final1-1lawrence hoÎncă nu există evaluări
- Des Met Ritual EnemasDocument280 paginiDes Met Ritual EnemascambiopacificoÎncă nu există evaluări
- Arvo Part ThesisDocument4 paginiArvo Part ThesisRX1986Încă nu există evaluări
- Hallberg Jonas A Randomized Controlled Study of Group 2019Document13 paginiHallberg Jonas A Randomized Controlled Study of Group 2019Sergio De PanfilisÎncă nu există evaluări
- An Analysis of Housing Conditions in AmacDocument38 paginiAn Analysis of Housing Conditions in AmacGodswill EssienÎncă nu există evaluări
- Jurnal 4Document13 paginiJurnal 4septi arum pradanaÎncă nu există evaluări
- Factors Affecting Students' Decision To Drop Out of School: Hermogenes C. Orion, JRDocument16 paginiFactors Affecting Students' Decision To Drop Out of School: Hermogenes C. Orion, JRJerico riveraÎncă nu există evaluări
- ORSC 341-Human Resource Management - Nabeel ArifDocument5 paginiORSC 341-Human Resource Management - Nabeel ArifSalma AzizÎncă nu există evaluări
- Exchange Variables As Predictors of Job Satisfaction, Job Commitment, and Turnover: The Impact of Rewards, Costs, Alternatives, and InvestmentsDocument18 paginiExchange Variables As Predictors of Job Satisfaction, Job Commitment, and Turnover: The Impact of Rewards, Costs, Alternatives, and InvestmentsPedro Alberto Herrera LedesmaÎncă nu există evaluări
- English GrammarDocument30 paginiEnglish GrammarBelén Pérez DíazÎncă nu există evaluări
- Can Perception Training Improve The Production of Second Language Phonemes? A Meta-Analytic Review of 25 Years of Perception Training ResearchDocument38 paginiCan Perception Training Improve The Production of Second Language Phonemes? A Meta-Analytic Review of 25 Years of Perception Training ResearchCarolina RochaÎncă nu există evaluări
- Fredda - Lesson PlanDocument6 paginiFredda - Lesson PlanLhinever GilhangÎncă nu există evaluări
- Soyoung CVDocument6 paginiSoyoung CVapi-477306025Încă nu există evaluări
- Group Behavior, Teams and ConflictDocument78 paginiGroup Behavior, Teams and ConflictEhla Silverio-Vito100% (1)
- Engineering Research Methodology Test #2 Part A: Question BankDocument2 paginiEngineering Research Methodology Test #2 Part A: Question BankrdsrajÎncă nu există evaluări
- An Effect of Dietary Supplementation of Chlorella: Vulgaris (Green Microalgae) On Egg ProductionDocument6 paginiAn Effect of Dietary Supplementation of Chlorella: Vulgaris (Green Microalgae) On Egg ProductionTJPRC PublicationsÎncă nu există evaluări
- Chap 1 The Nature of Management Control SystemsDocument12 paginiChap 1 The Nature of Management Control SystemsShaeesta ChowdharyÎncă nu există evaluări
- Introduction To SPSSDocument17 paginiIntroduction To SPSSUrba AshfaqÎncă nu există evaluări
- Ebook STNAR 02rev1 EDocument138 paginiEbook STNAR 02rev1 Ejohnn criolloÎncă nu există evaluări
- Chapter 4 (AZOLLA)Document8 paginiChapter 4 (AZOLLA)Jomari TawatÎncă nu există evaluări
- Stanford Stats 200Document6 paginiStanford Stats 200Jung Yoon SongÎncă nu există evaluări
- Zelazo Et Al 2024 Fostering Executive Function Skills and Promoting Far Transfer To Real World Outcomes The ImportanceDocument7 paginiZelazo Et Al 2024 Fostering Executive Function Skills and Promoting Far Transfer To Real World Outcomes The ImportanceultimatedeliciaÎncă nu există evaluări