S-ar putea să vă placă și
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- Timothy Dwight - Theology Explained and Defended - Series of SermonsDocument648 paginiTimothy Dwight - Theology Explained and Defended - Series of SermonsPaolopiniÎncă nu există evaluări
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- Pyramids - Ground Penetrating RadarDocument16 paginiPyramids - Ground Penetrating RadarBryan GraczykÎncă nu există evaluări
- Multisim 12 - Flip FlopDocument6 paginiMultisim 12 - Flip FlopPaolopiniÎncă nu există evaluări
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Khavrsohkin - Geophysical Fields Around The PyramidsDocument41 paginiKhavrsohkin - Geophysical Fields Around The PyramidsPaolopiniÎncă nu există evaluări
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Timothy Dwight - Theology Explained and Defended - Series of SermonsDocument648 paginiTimothy Dwight - Theology Explained and Defended - Series of SermonsPaolopiniÎncă nu există evaluări
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- Maldacena Juan - The Large N Limit of Superconformal Eld Theories and Supergravity 9711200 PDFDocument22 paginiMaldacena Juan - The Large N Limit of Superconformal Eld Theories and Supergravity 9711200 PDFPaolopiniÎncă nu există evaluări
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (399)
- Italian Society of Neurology Italo-Swiss MeetingDocument1 paginăItalian Society of Neurology Italo-Swiss MeetingPaolopiniÎncă nu există evaluări
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- Italian Society of Neurology Italo-Swiss MeetingDocument1 paginăItalian Society of Neurology Italo-Swiss MeetingPaolopiniÎncă nu există evaluări
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- Beyond The Material UniverseDocument25 paginiBeyond The Material UniversePaolopiniÎncă nu există evaluări
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- The Parkin Gene and Its PhenotypeDocument2 paginiThe Parkin Gene and Its PhenotypePaolopiniÎncă nu există evaluări
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Usefulness of The Italian Version of Parkinson's Disease Sleep ScaleDocument6 paginiUsefulness of The Italian Version of Parkinson's Disease Sleep ScalePaolopiniÎncă nu există evaluări
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Tarzan Chronology2Document12 paginiTarzan Chronology2PaolopiniÎncă nu există evaluări
- The Parkin Gene and Its PhenotypeDocument2 paginiThe Parkin Gene and Its PhenotypePaolopiniÎncă nu există evaluări
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (73)
- Psychiatric Symptoms in Parkinson's DiseaseDocument1 paginăPsychiatric Symptoms in Parkinson's DiseasePaolopiniÎncă nu există evaluări
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- The Parkin Gene and Its PhenotypeDocument2 paginiThe Parkin Gene and Its PhenotypePaolopiniÎncă nu există evaluări
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- Neurite Outgrowth Assay NEODocument2 paginiNeurite Outgrowth Assay NEOPaolopiniÎncă nu există evaluări
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- Psychiatric Symptoms in Parkinson's DiseaseDocument1 paginăPsychiatric Symptoms in Parkinson's DiseasePaolopiniÎncă nu există evaluări
- Speciale - Terapia Crescita Dei NeuritiDocument1 paginăSpeciale - Terapia Crescita Dei NeuritiPaolopiniÎncă nu există evaluări
- Lifestyle-Related Risk Factors For Parkinson's Disease - A Population-Based StudyDocument6 paginiLifestyle-Related Risk Factors For Parkinson's Disease - A Population-Based StudyPaolopiniÎncă nu există evaluări
- Occupation, Education, and Parkinson's Disease - A Case-Control Study in An Italian PopulationDocument6 paginiOccupation, Education, and Parkinson's Disease - A Case-Control Study in An Italian PopulationPaolopiniÎncă nu există evaluări
- Latex-Lyx (Amir Karger) (ITA)Document41 paginiLatex-Lyx (Amir Karger) (ITA)PaolopiniÎncă nu există evaluări
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- Lifestyle-Related Risk Factors For Parkinson's Disease - A Population-Based StudyDocument6 paginiLifestyle-Related Risk Factors For Parkinson's Disease - A Population-Based StudyPaolopiniÎncă nu există evaluări
- Introduction To Te XDocument97 paginiIntroduction To Te XClio NapoliÎncă nu există evaluări
- Usefulness of The Italian Version of Parkinson's Disease Sleep ScaleDocument6 paginiUsefulness of The Italian Version of Parkinson's Disease Sleep ScalePaolopiniÎncă nu există evaluări
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- RCD ManagementDocument6 paginiRCD ManagementPindoterOÎncă nu există evaluări
- SalerioDocument28 paginiSalerioRizqaFebrilianyÎncă nu există evaluări
- CA-idms Ads Alive User Guide 15.0Document142 paginiCA-idms Ads Alive User Guide 15.0svdonthaÎncă nu există evaluări
- Rotc Reviewer FinalsDocument11 paginiRotc Reviewer FinalsAngel Atienza100% (1)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (120)
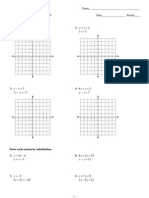
- Review Systems of Linear Equations All MethodsDocument4 paginiReview Systems of Linear Equations All Methodsapi-265647260Încă nu există evaluări
- Assignment Group OSHADocument10 paginiAssignment Group OSHAariffikriismailÎncă nu există evaluări
- Architech 06-2016 Room AssignmentDocument4 paginiArchitech 06-2016 Room AssignmentPRC Baguio100% (1)
- Keira Knightley: Jump To Navigation Jump To SearchDocument12 paginiKeira Knightley: Jump To Navigation Jump To SearchCrina LupuÎncă nu există evaluări
- UntitledDocument17 paginiUntitledSedat100% (1)
- Evolution of Strategic HRM As Seen Through Two Founding Books A 30TH Anniversary Perspective On Development of The FieldDocument20 paginiEvolution of Strategic HRM As Seen Through Two Founding Books A 30TH Anniversary Perspective On Development of The FieldJhon Alex ValenciaÎncă nu există evaluări
- Risk Assessment For ExcavationDocument6 paginiRisk Assessment For ExcavationAhmed GamalÎncă nu există evaluări
- Automatic Coconut Dehusking MachineDocument12 paginiAutomatic Coconut Dehusking MachineKumaresh Salem0% (1)
- Loch ChildrenDocument4 paginiLoch ChildrenLauro De Jesus FernandesÎncă nu există evaluări
- Architecture of Neural NWDocument79 paginiArchitecture of Neural NWapi-3798769Încă nu există evaluări
- About TableauDocument22 paginiAbout TableauTarun Sharma67% (3)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- Project Proposal DraftDocument1 paginăProject Proposal DraftCarl Axel M. FajardoÎncă nu există evaluări
- A Review On Different Yogas Used in The Management of Mandali Damsa Vrana W.S.R. To KriyakaumudiDocument11 paginiA Review On Different Yogas Used in The Management of Mandali Damsa Vrana W.S.R. To KriyakaumudiTiya TiwariÎncă nu există evaluări
- Data Analyst Chapter 3Document20 paginiData Analyst Chapter 3Andi Annisa DianputriÎncă nu există evaluări
- Exam C - HANATEC142: SAP Certified Technology Associate - SAP HANA (Edition 2014)Document10 paginiExam C - HANATEC142: SAP Certified Technology Associate - SAP HANA (Edition 2014)SadishÎncă nu există evaluări
- AR Financial StatementsDocument281 paginiAR Financial StatementsISHA AGGARWALÎncă nu există evaluări
- Business EthicsDocument10 paginiBusiness EthicsTeguh HardiÎncă nu există evaluări
- Neuroscience Core ConceptsDocument2 paginiNeuroscience Core Conceptseglantina alishollariÎncă nu există evaluări
- Avanquest Perfect Image V.12 User GuideDocument174 paginiAvanquest Perfect Image V.12 User GuideShafiq-UR-Rehman Lodhi100% (1)
- MELASMA (Ardy, Kintan, Fransisca)Document20 paginiMELASMA (Ardy, Kintan, Fransisca)Andi Firman MubarakÎncă nu există evaluări
- AC AMMETER / Moving Iron: Model AECDocument33 paginiAC AMMETER / Moving Iron: Model AECRoonar Aponte NoaÎncă nu există evaluări
- Grammar Review A2-B1Document5 paginiGrammar Review A2-B1Lena Silva SouzaÎncă nu există evaluări
- Episode Transcript: Episode 34 - Chinese Han Lacquer CupDocument2 paginiEpisode Transcript: Episode 34 - Chinese Han Lacquer CupParvathy SubramanianÎncă nu există evaluări
- UV-Visible Systems - Operational Qualification - Col23 PDFDocument10 paginiUV-Visible Systems - Operational Qualification - Col23 PDFIsabelle PlourdeÎncă nu există evaluări
- Sharp Product-Catalogue 2019 enDocument48 paginiSharp Product-Catalogue 2019 enMiki di KaprioÎncă nu există evaluări
- L&T Motor CatalogDocument24 paginiL&T Motor CatalogSanjeev DhariwalÎncă nu există evaluări
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeDe la EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeEvaluare: 2 din 5 stele2/5 (1)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsDe la EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsÎncă nu există evaluări
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisDe la EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisEvaluare: 4.5 din 5 stele4.5/5 (42)
- The Age of Magical Overthinking: Notes on Modern IrrationalityDe la EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityEvaluare: 4 din 5 stele4/5 (24)