S-ar putea să vă placă și
- Hirschsprung's Disease: (Congenital Aganglionic Megacolon)Document16 paginiHirschsprung's Disease: (Congenital Aganglionic Megacolon)umiÎncă nu există evaluări
- Hischsprug Disease by HamadDocument31 paginiHischsprug Disease by HamadALIEF MUTHIAÎncă nu există evaluări
- Hirschsprung Disease Case Study: Maecy P. Tarinay BSN 4-1Document5 paginiHirschsprung Disease Case Study: Maecy P. Tarinay BSN 4-1Maecy OdegaardÎncă nu există evaluări
- Hirschsprung DiseaseDocument44 paginiHirschsprung DiseaseAhmad Abu KushÎncă nu există evaluări
- Congenital Aganglionic Megacolon (Hirschsprung Disease) : Kristin N. Fiorino and Chris A. LiacourasDocument6 paginiCongenital Aganglionic Megacolon (Hirschsprung Disease) : Kristin N. Fiorino and Chris A. LiacourasSyakilla AuliaÎncă nu există evaluări
- ILA - Hirschsprungs DiseaseDocument48 paginiILA - Hirschsprungs DiseaseSoleh Ramly100% (1)
- Hirschsprung's Disease PathophysiologyDocument8 paginiHirschsprung's Disease Pathophysiologyclaire yowsÎncă nu există evaluări
- Hirschprung's DiseaseDocument26 paginiHirschprung's DiseaseAbdur RaqibÎncă nu există evaluări
- Congenital Aganglionic Megacolon - Hirschsprung Disease - 2009-6Document49 paginiCongenital Aganglionic Megacolon - Hirschsprung Disease - 2009-6Muhammad SubhanÎncă nu există evaluări
- Neonatal Intestinal Obstruction EPSGHAN PDFDocument77 paginiNeonatal Intestinal Obstruction EPSGHAN PDFRobert ChristevenÎncă nu există evaluări
- Hirschsprung 1socaDocument34 paginiHirschsprung 1socaDianÎncă nu există evaluări
- Hisprung DiseaseDocument12 paginiHisprung DiseaseEky Madyaning NastitiÎncă nu există evaluări
- Hirschsprung DiseaseDocument25 paginiHirschsprung DiseaseMuhammad Zaniar RamadhaniÎncă nu există evaluări
- Intussuseption and Hirschprung's DiseaseDocument5 paginiIntussuseption and Hirschprung's DiseaseAris Magallanes100% (2)
- GERD in ChildrenDocument31 paginiGERD in ChildrenSalman KhanÎncă nu există evaluări
- Hirschsprung Disease: Nadia Ismael Muse Safia Ahmed-Yassin SH: Ali Ilham Saed JirdeDocument24 paginiHirschsprung Disease: Nadia Ismael Muse Safia Ahmed-Yassin SH: Ali Ilham Saed Jirdesafia ahmedÎncă nu există evaluări
- Biliary atresia, Hirschsprung's disease, and anorectal malformationsDocument4 paginiBiliary atresia, Hirschsprung's disease, and anorectal malformationsMohamed Al-zichrawyÎncă nu există evaluări
- Hirschsprung DiseaseDocument20 paginiHirschsprung DiseaseIvy DanÎncă nu există evaluări
- Constipation in ChildrenDocument34 paginiConstipation in Childrenabdisalaan hassanÎncă nu există evaluări
- Prune Belly SyndromeDocument39 paginiPrune Belly SyndromeHudaÎncă nu există evaluări
- Hirsch SprungDocument20 paginiHirsch SprungrisaÎncă nu există evaluări
- Intestinal Pathology III Hirschsprung'S Disease: DR Nzau MuangeDocument21 paginiIntestinal Pathology III Hirschsprung'S Disease: DR Nzau MuangeNzau MuangeÎncă nu există evaluări
- Hirschsprun G'S Disease: Dr. Manish Kumar Gupta Assistant Professor Department of Paediatric Surgery AIIMS, RishikeshDocument48 paginiHirschsprun G'S Disease: Dr. Manish Kumar Gupta Assistant Professor Department of Paediatric Surgery AIIMS, RishikeshArchana Mahata100% (1)
- HirschprungDocument6 paginiHirschprungVanessa CasingalÎncă nu există evaluări
- Hirschsprung Disease: Waardenburg Syndrome Mowat-Wilson Syndrome Congenital Central Hypoventilation SyndromeDocument22 paginiHirschsprung Disease: Waardenburg Syndrome Mowat-Wilson Syndrome Congenital Central Hypoventilation SyndromeRizki Nandasari SulbahriÎncă nu există evaluări
- Lec 1Document52 paginiLec 1zainabd1964Încă nu există evaluări
- Necrotizing EnterocolitisDocument36 paginiNecrotizing EnterocolitisMahad Maxamed AxmedÎncă nu există evaluări
- Lecture 1 HirschprungDocument18 paginiLecture 1 HirschprungsharmeenÎncă nu există evaluări
- Congenital AnomaliesDocument10 paginiCongenital Anomaliesربيع ضياء ربيعÎncă nu există evaluări
- Askep HisprungDocument25 paginiAskep HisprungRika AmaliyaÎncă nu există evaluări
- No Bowel Output in NeonatesDocument24 paginiNo Bowel Output in NeonatesOTOH RAYA OMARÎncă nu există evaluări
- Hirschsprung's Disease ExplainedDocument11 paginiHirschsprung's Disease ExplainedKarl JoseÎncă nu există evaluări
- Hirschsprung DiseaseDocument17 paginiHirschsprung DiseaseJesselyn Heruela100% (1)
- Hirsch SprungDocument16 paginiHirsch SprungjessyÎncă nu există evaluări
- 12 - Paediatric Abdomen RadiologyDocument74 pagini12 - Paediatric Abdomen RadiologyMaria DoukaÎncă nu există evaluări
- Intussusception PresentationDocument9 paginiIntussusception PresentationAme NasokiaÎncă nu există evaluări
- ConstipationDocument33 paginiConstipationsalmawalidÎncă nu există evaluări
- Hirschsprung DiseaseDocument19 paginiHirschsprung DiseaseUgi Rahul100% (1)
- Chapter X.4. Intussusception: Case Based Pediatrics For Medical Students and ResidentsDocument5 paginiChapter X.4. Intussusception: Case Based Pediatrics For Medical Students and ResidentsNawaf Rahi AlshammariÎncă nu există evaluări
- Problem 4 Git: Ivan Michael (405090161)Document22 paginiProblem 4 Git: Ivan Michael (405090161)vnÎncă nu există evaluări
- 0327 Congenital Anomalies Rutter Congenital Anomalies Tracheoesophageal Fistula and Esophageal AtresiaDocument6 pagini0327 Congenital Anomalies Rutter Congenital Anomalies Tracheoesophageal Fistula and Esophageal AtresiaYvonne ChuehÎncă nu există evaluări
- Hirschsprung Disease (Aganglionic Megacolon)Document6 paginiHirschsprung Disease (Aganglionic Megacolon)Julliza Joy PandiÎncă nu există evaluări
- PEDIATRIC SURGERY PROBLEMSDocument141 paginiPEDIATRIC SURGERY PROBLEMSsedaka26100% (4)
- Hirschsprung Disease FarieDocument38 paginiHirschsprung Disease FarieFarie Farihan67% (3)
- Malformations of OesophagusDocument24 paginiMalformations of OesophagusOwoupele Ak-Gabriel100% (1)
- Pediatric Clinics of North America IIDocument54 paginiPediatric Clinics of North America IIkarenÎncă nu există evaluări
- Key signs and symptoms of infantile pyloric stenosisDocument6 paginiKey signs and symptoms of infantile pyloric stenosisNeil AlviarÎncă nu există evaluări
- Hirschsprung DiseaseDocument21 paginiHirschsprung DiseaseAhmad YaniÎncă nu există evaluări
- Gastric DisordersDocument135 paginiGastric DisordersEsmareldah Henry SirueÎncă nu există evaluări
- Disorders of The IntestinesDocument64 paginiDisorders of The IntestinesCharmaine Torio PastorÎncă nu există evaluări
- Hirschsprung DiseaseDocument1 paginăHirschsprung DiseasePutri ClaraÎncă nu există evaluări
- Hirschsprung'S Disease: DR: Jose Antonio Hernandez LivenDocument32 paginiHirschsprung'S Disease: DR: Jose Antonio Hernandez Livenmomodou s jallowÎncă nu există evaluări
- Emergencies in GITDocument7 paginiEmergencies in GITsssajiÎncă nu există evaluări
- Hirschsprung Disease: Causes, Symptoms and Treatment/TITLEDocument61 paginiHirschsprung Disease: Causes, Symptoms and Treatment/TITLEAdditi Satyal100% (1)
- Esophageal diseases and surgical managementDocument72 paginiEsophageal diseases and surgical managementBiniamÎncă nu există evaluări
- Enfermedad de Hirschsprung: Diagnóstico Y Manejo en Niños Y AdultosDocument5 paginiEnfermedad de Hirschsprung: Diagnóstico Y Manejo en Niños Y AdultosAlexander Castillo CalderónÎncă nu există evaluări
- Sick Growing Child: Hirschsprung Disease Anorectal Malformation Maluenda CamilleDocument46 paginiSick Growing Child: Hirschsprung Disease Anorectal Malformation Maluenda CamilleCamille Maluenda - TanÎncă nu există evaluări
- SG3 Paediatric Surgical EmergenciesDocument69 paginiSG3 Paediatric Surgical EmergenciesDiyana ZatyÎncă nu există evaluări
- Dysphagia, A Simple Guide To The Condition, Treatment And Related ConditionsDe la EverandDysphagia, A Simple Guide To The Condition, Treatment And Related ConditionsEvaluare: 5 din 5 stele5/5 (1)
- PRC Form Cmo 14 IrishDocument6 paginiPRC Form Cmo 14 IrishJonathan ObañaÎncă nu există evaluări
- Mnemonic DeviceDocument12 paginiMnemonic DeviceJonathan ObañaÎncă nu există evaluări
- Assessing pedal pulses in patients with hip fracturesDocument5 paginiAssessing pedal pulses in patients with hip fracturesJonathan ObañaÎncă nu există evaluări
- Statistics and its role in data analysis, interpretation and decision makingDocument1 paginăStatistics and its role in data analysis, interpretation and decision makingJonathan ObañaÎncă nu există evaluări
- Alkylating AgentsDocument3 paginiAlkylating AgentsJonathan ObañaÎncă nu există evaluări
- Plan of ActivityDocument2 paginiPlan of ActivityJonathan ObañaÎncă nu există evaluări
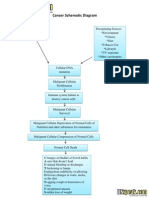
- Cancer Schematic DiagramDocument1 paginăCancer Schematic DiagramJonathan ObañaÎncă nu există evaluări
- Herniated Nucleus PulposusDocument4 paginiHerniated Nucleus PulposusJonathan ObañaÎncă nu există evaluări
- In IvfusionDocument5 paginiIn IvfusionJonathan ObañaÎncă nu există evaluări
- Medifocus April 2007Document61 paginiMedifocus April 2007Pushpanjali Crosslay HospitalÎncă nu există evaluări
- Module 1: Disability, Inequality, and InclusionDocument25 paginiModule 1: Disability, Inequality, and InclusionAuberon Jeleel OdoomÎncă nu există evaluări
- Lab Result - 742124389Document1 paginăLab Result - 742124389Shivanshu RajputÎncă nu există evaluări
- Tle ActivityDocument4 paginiTle ActivityManayam CatherineÎncă nu există evaluări
- FACULTY OF APPLIED SCIENCE BIO320 CASE STUDY: LEPTOSPIROSIS IN MALAYSIADocument12 paginiFACULTY OF APPLIED SCIENCE BIO320 CASE STUDY: LEPTOSPIROSIS IN MALAYSIAIlham Amni AmaninaÎncă nu există evaluări
- Lab Report: 2743025 LAB/20N/121831 27/jan/2022 Mr. Naman Thapliyal 13649512 StatusDocument2 paginiLab Report: 2743025 LAB/20N/121831 27/jan/2022 Mr. Naman Thapliyal 13649512 StatusM Abdul MoidÎncă nu există evaluări
- Purified Used Cooking Oil Through SedimentationDocument4 paginiPurified Used Cooking Oil Through SedimentationJan Elison NatividadÎncă nu există evaluări
- Minnesota Multiphasic Personality Inventory TestDocument26 paginiMinnesota Multiphasic Personality Inventory TestGicu BelicuÎncă nu există evaluări
- Establishing Positive Family Relationships for Health PromotionDocument78 paginiEstablishing Positive Family Relationships for Health PromotionsunielgowdaÎncă nu există evaluări
- Digital Rectal ExaminationDocument7 paginiDigital Rectal ExaminationAla'a Emerald AguamÎncă nu există evaluări
- Ovitrelle Epar Medicine Overview - enDocument3 paginiOvitrelle Epar Medicine Overview - enPhysics with V SagarÎncă nu există evaluări
- Safety Data Sheet: 1. Identification of The Substance / Preparation and of The Company / UndertakingDocument3 paginiSafety Data Sheet: 1. Identification of The Substance / Preparation and of The Company / UndertakingnbagarÎncă nu există evaluări
- Vdoc - Pub End of Life Care in Cardiovascular DiseaseDocument251 paginiVdoc - Pub End of Life Care in Cardiovascular Diseasericardo valtierra diaz infanteÎncă nu există evaluări
- Vaccination Program in Farm AnimalDocument87 paginiVaccination Program in Farm AnimalThành Đỗ MinhÎncă nu există evaluări
- 4th Yr SingiDocument322 pagini4th Yr SingiSthuti Shetwal100% (1)
- The Nature and Meaning of Physical EducationDocument14 paginiThe Nature and Meaning of Physical EducationRegie Mark MansigueÎncă nu există evaluări
- Soal Lokasi KIK - 2Document2 paginiSoal Lokasi KIK - 2novida nainggolanÎncă nu există evaluări
- Aunt Minnie Pediatric NeuroDocument15 paginiAunt Minnie Pediatric NeuroRommel OliverasÎncă nu există evaluări
- Impact of Small-Scale Mining To The ResidentsDocument15 paginiImpact of Small-Scale Mining To The ResidentsAngelIgualdoSagapiÎncă nu există evaluări
- Patient Examination: History: by Professor of Internal MedicineDocument47 paginiPatient Examination: History: by Professor of Internal MedicineMonqith YousifÎncă nu există evaluări
- ANM application detailsDocument3 paginiANM application detailsSOFIKUL HUUSAINÎncă nu există evaluări
- Southampton Grading SystemDocument5 paginiSouthampton Grading SystemswestyÎncă nu există evaluări
- Herboboost Capsules: Kutki, Chandan)Document3 paginiHerboboost Capsules: Kutki, Chandan)Snehal AshtekarÎncă nu există evaluări
- CMC Vellore Summer Admission Bulletin 2020 Revised 16 Nov 2020Document58 paginiCMC Vellore Summer Admission Bulletin 2020 Revised 16 Nov 2020Allen ChrysoÎncă nu există evaluări
- Peginesatide For Anemia in Patients With Chronic Kidney Disease Not Receiving DialysisDocument21 paginiPeginesatide For Anemia in Patients With Chronic Kidney Disease Not Receiving DialysisEdi Kurnawan TjhaiÎncă nu există evaluări
- Effects of Drug Abuse on Health and SocietyDocument9 paginiEffects of Drug Abuse on Health and SocietyAditya guptaÎncă nu există evaluări
- E-Learning PHARM 131 - Chapter2Document10 paginiE-Learning PHARM 131 - Chapter2Ryan Charles Uminga GalizaÎncă nu există evaluări
- Winters Ildp Final Review Draft ApprovedDocument7 paginiWinters Ildp Final Review Draft Approvedapi-424954609Încă nu există evaluări
- PNLE June 2007 With Key AnswersDocument82 paginiPNLE June 2007 With Key AnswersJustin CubillasÎncă nu există evaluări
- LastCallProgram ScienceDocument16 paginiLastCallProgram ScienceConiforÎncă nu există evaluări