S-ar putea să vă placă și
- Object 1: Easy German BookDocument4 paginiObject 1: Easy German BookMelina Student - ChatzisymeonÎncă nu există evaluări
- Biology IADocument16 paginiBiology IAAngelina TomacÎncă nu există evaluări
- Evaluate Techniques Used To Study The Brain in Relation To Human Behavior.Document3 paginiEvaluate Techniques Used To Study The Brain in Relation To Human Behavior.Ashley LauÎncă nu există evaluări
- 2012 Probability Past IB QuestionsDocument18 pagini2012 Probability Past IB QuestionsVictor O. WijayaÎncă nu există evaluări
- ToK Journal - ImaginationDocument2 paginiToK Journal - ImaginationInundatedU100% (1)
- IB-Biology-SL Ocs Revision GuideDocument1 paginăIB-Biology-SL Ocs Revision Guidetg206Încă nu există evaluări
- Chemistry Extended EssayDocument45 paginiChemistry Extended EssayYang HaoÎncă nu există evaluări
- Introduction To Cells (1.1) :: The Cell TheoryDocument65 paginiIntroduction To Cells (1.1) :: The Cell Theoryelvira sta.mariaÎncă nu există evaluări
- Cas EssayDocument2 paginiCas EssaySara AbouhilalÎncă nu există evaluări
- Honors Chemistry Study Guide For Semester I Final ExamDocument2 paginiHonors Chemistry Study Guide For Semester I Final ExamBilal QureshiÎncă nu există evaluări
- IB Final Exam NotesDocument22 paginiIB Final Exam NotesJian Zhi TehÎncă nu există evaluări
- Human NutritionDocument53 paginiHuman NutritionTanyaÎncă nu există evaluări
- SOcial Cognitive TheoryDocument5 paginiSOcial Cognitive TheoryKritarth BhasinÎncă nu există evaluări
- Grade 11 Biology Midterm HLDocument31 paginiGrade 11 Biology Midterm HLSaima SyedaÎncă nu există evaluări
- June 2019 (IAL) QP - Unit 4 Edexcel Economics A-LevelDocument44 paginiJune 2019 (IAL) QP - Unit 4 Edexcel Economics A-LevelFahad FarhanÎncă nu există evaluări
- Biology Extended Essay Final Draft.Document33 paginiBiology Extended Essay Final Draft.Oliver Hernández100% (1)
- IB Chemistry Lab Report ChecklistDocument3 paginiIB Chemistry Lab Report ChecklistoanaivnÎncă nu există evaluări
- 43ac5f1d 4d98 4a14 8a5f C110b7e4ee79 - Psychology Edexcel WorkbookDocument141 pagini43ac5f1d 4d98 4a14 8a5f C110b7e4ee79 - Psychology Edexcel WorkbookShawn VargheseÎncă nu există evaluări
- Biology Extended EssayDocument8 paginiBiology Extended Essaya100% (1)
- 1.4 Membrane Transport (53 Marks) : MarkschemeDocument20 pagini1.4 Membrane Transport (53 Marks) : Markschemelayal hashem0% (2)
- IB Biology Extended Essay - A ScoringDocument36 paginiIB Biology Extended Essay - A ScoringCaitlinÎncă nu există evaluări
- Biology HL FlashcardsDocument22 paginiBiology HL FlashcardsTiegan Blake100% (1)
- Biology AQA Chapter 1 Questions and AnswersDocument18 paginiBiology AQA Chapter 1 Questions and Answersmahamed100% (1)
- Topic 2 - Mechanics - IB PhysicsDocument23 paginiTopic 2 - Mechanics - IB PhysicsAzzahra Yeasmin Saika100% (1)
- CHPT 15 Genetics Practice ProblemsDocument7 paginiCHPT 15 Genetics Practice ProblemsFatimah AliyaÎncă nu există evaluări
- Topic 1.1 - Measurements in PhysicsDocument46 paginiTopic 1.1 - Measurements in PhysicsPaul Amezquita100% (1)
- IA Chemistry 1Document13 paginiIA Chemistry 1priyaÎncă nu există evaluări
- Oxidation Reduction Past PaperDocument8 paginiOxidation Reduction Past Paperrainbow100% (1)
- Essay Plan TOKDocument3 paginiEssay Plan TOKSimran Toor0% (3)
- IB Bio IA On Nuruk FermentationDocument9 paginiIB Bio IA On Nuruk FermentationGoeun Jeong (yr. 18-20)Încă nu există evaluări
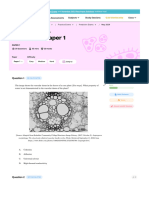
- IB Biology SL - 2024 Prediction Exam - May 2024 Paper 1Document16 paginiIB Biology SL - 2024 Prediction Exam - May 2024 Paper 1Christy HuynhÎncă nu există evaluări
- TOK EssayDocument3 paginiTOK EssayOlivier SorghoÎncă nu există evaluări
- Bio Mini IA Design (HL)Document7 paginiBio Mini IA Design (HL)Lorraine VictoriaÎncă nu există evaluări
- Physics Extended Essay PDFDocument5 paginiPhysics Extended Essay PDFDiyaÎncă nu există evaluări
- IB Biology Lab ReportDocument21 paginiIB Biology Lab ReportAgnieszka100% (1)
- Internal Assesment Biology - 25Document11 paginiInternal Assesment Biology - 25lauraÎncă nu există evaluări
- Transcript of IB Chemistry Mind MapDocument8 paginiTranscript of IB Chemistry Mind MapJayakumar SankaranÎncă nu există evaluări
- Ib Extended Essay in BiologyDocument2 paginiIb Extended Essay in Biologyapi-261035067Încă nu există evaluări
- #2 ERQ - OutlineDocument3 pagini#2 ERQ - OutlineEvergreen EvanyÎncă nu există evaluări
- IB HL BIO FULL NOTES (Onarıldı)Document874 paginiIB HL BIO FULL NOTES (Onarıldı)Sıla DenizÎncă nu există evaluări
- Lab Report Guidelines For Ib in BiologyDocument5 paginiLab Report Guidelines For Ib in BiologyCheyenne AlvaradoÎncă nu există evaluări
- Ib Chemistry SL BookletDocument8 paginiIb Chemistry SL BookletBoshra NouriÎncă nu există evaluări
- Chemical KineticsDocument7 paginiChemical KineticsdineshnpÎncă nu există evaluări
- Ia FinalDocument20 paginiIa Finalapi-253984189Încă nu există evaluări
- Markscheme: (125 Marks)Document33 paginiMarkscheme: (125 Marks)Naz GümüşlüoğluÎncă nu există evaluări
- Rubric Ee Visual ArtsDocument5 paginiRubric Ee Visual Artsapi-307142321Încă nu există evaluări
- Topic 7 Practice Paper 2 IB ChemistryDocument6 paginiTopic 7 Practice Paper 2 IB ChemistryDea SukrisnaÎncă nu există evaluări
- Tok EssayDocument7 paginiTok Essayapi-648772994Încă nu există evaluări
- SL Biology Syllabus NotesDocument52 paginiSL Biology Syllabus NotesRyel MuchunkuÎncă nu există evaluări
- GUIDE To The IA in IB BiologyDocument14 paginiGUIDE To The IA in IB BiologyJohn Osborne100% (1)
- Practice Questions Topic 1.4 IB BiologyDocument12 paginiPractice Questions Topic 1.4 IB BiologySamantha DAIROÎncă nu există evaluări
- Exceptions To The Cell TheoryDocument5 paginiExceptions To The Cell Theoryapi-326952599Încă nu există evaluări
- Bio IADocument13 paginiBio IAALEXANDRIA JEAN BURROUGHESÎncă nu există evaluări
- Periodic Table NotesDocument10 paginiPeriodic Table Notesapi-293690555Încă nu există evaluări
- Are Some Types of Knowledge More Useful Than Others?Document5 paginiAre Some Types of Knowledge More Useful Than Others?체르니30Încă nu există evaluări
- IB Psychology A Student's Guide Preview 3Document25 paginiIB Psychology A Student's Guide Preview 3Samiya SahuÎncă nu există evaluări
- IB Psychology ERQsDocument15 paginiIB Psychology ERQsMichelle Karam LondoñoÎncă nu există evaluări
- Biology Notes IB Free ResponseDocument26 paginiBiology Notes IB Free ResponseLarry LohÎncă nu există evaluări
- PCR Induction Day Destination WonderDocument6 paginiPCR Induction Day Destination Wonderapi-245190275Încă nu există evaluări
- NanotitrationDocument2 paginiNanotitrationapi-245190275Încă nu există evaluări
- Extended Essay Chemistry New Version 2Document40 paginiExtended Essay Chemistry New Version 2api-245190275100% (12)
- Ib Extended Essay Final - FinalDocument28 paginiIb Extended Essay Final - Finalapi-245190275Încă nu există evaluări
- Pseudo Pelger Huet Anomaly Induced by MedicationsDocument13 paginiPseudo Pelger Huet Anomaly Induced by MedicationsHam BoneÎncă nu există evaluări
- PCR Mycoplasma Test Kit I/C: Instruction ManualDocument8 paginiPCR Mycoplasma Test Kit I/C: Instruction ManualAslam MikraniÎncă nu există evaluări
- Real-Time PCR: CFX96 SystemDocument8 paginiReal-Time PCR: CFX96 SystemyÎncă nu există evaluări
- BAM Chapter 29. CronobacterDocument14 paginiBAM Chapter 29. CronobacterremyÎncă nu există evaluări
- 9-Diagnosis of Malaria InfectionDocument27 pagini9-Diagnosis of Malaria InfectionMazterMaztermaztermazter MaztermaztermazterYandeÎncă nu există evaluări
- Iso 20395Document44 paginiIso 20395Gilberto Ragnar Nodarse100% (1)
- 04 Plant Taxonomy ManualDocument56 pagini04 Plant Taxonomy ManualCapale MCÎncă nu există evaluări
- Lewis Et Al-2001-The Journal of PathologyDocument6 paginiLewis Et Al-2001-The Journal of PathologyHaekal HafizhÎncă nu există evaluări
- Molecular Epidemiologi CancerDocument10 paginiMolecular Epidemiologi CancerM NnÎncă nu există evaluări
- Extraction of DNA From Whole BloodDocument5 paginiExtraction of DNA From Whole BloodvishankguptaÎncă nu există evaluări
- Isothermal Nucleic Acid Amplification For Point of Care DevicesDocument27 paginiIsothermal Nucleic Acid Amplification For Point of Care DevicespascalcrawÎncă nu există evaluări
- School of Mathematics and Natural Sciences: Irina - Borovkov@utdallas - EduDocument4 paginiSchool of Mathematics and Natural Sciences: Irina - Borovkov@utdallas - EdubillyÎncă nu există evaluări
- Fate of SRY, PABY, DYS1, DYZ3 and DYZ1 Loci in Indian Patients Harbouring Sex Chromosomal AnomaliesDocument11 paginiFate of SRY, PABY, DYS1, DYZ3 and DYZ1 Loci in Indian Patients Harbouring Sex Chromosomal Anomaliesrenny100% (1)
- BCSRC Researchpaper PDFDocument25 paginiBCSRC Researchpaper PDFapi-509442117Încă nu există evaluări
- Little Leaf of BrinjalDocument64 paginiLittle Leaf of BrinjalPARITOSH SHARMAÎncă nu există evaluări
- Isolation and Identification Ort From ChickenDocument14 paginiIsolation and Identification Ort From Chickenthanh ba matÎncă nu există evaluări
- Flaws in Coronavirus Pandemic Theory: 1. Executive SummaryDocument50 paginiFlaws in Coronavirus Pandemic Theory: 1. Executive SummaryWendell StantonÎncă nu există evaluări
- 258-265Document8 pagini258-265Muhilan MahendhiranÎncă nu există evaluări
- Powell Et Al. 2006 ShrimpleDocument6 paginiPowell Et Al. 2006 ShrimpleerinjburgeÎncă nu există evaluări
- Rev. Lect 2. MOLECULAR TECHNIQUES IN DIAGNOSTIC MICROBIOLOGYDocument75 paginiRev. Lect 2. MOLECULAR TECHNIQUES IN DIAGNOSTIC MICROBIOLOGYKhrys HardyÎncă nu există evaluări
- Research & Development: Test Name Status Result Unit Reference Interval SARS-COV-2 Real-Time PCR, QualitativeDocument2 paginiResearch & Development: Test Name Status Result Unit Reference Interval SARS-COV-2 Real-Time PCR, QualitativeakashÎncă nu există evaluări
- Biotechnology: Quarter 3 - Module 2: Genetic ManipulationDocument24 paginiBiotechnology: Quarter 3 - Module 2: Genetic ManipulationJohn Mark Laurio82% (17)
- KOD Hot StartDocument4 paginiKOD Hot Startsike1977Încă nu există evaluări
- QualityindicatorsDocument8 paginiQualityindicatorsMishalni PotterÎncă nu există evaluări
- Incompatibility and The Partitioning Site of The repABC Basic Replicon of The Symbiotic Plasmid From Rhizobium EtliDocument14 paginiIncompatibility and The Partitioning Site of The repABC Basic Replicon of The Symbiotic Plasmid From Rhizobium EtliMiguel RamirezÎncă nu există evaluări
- KQB 7002 Bioinstrumentation Case StudyDocument14 paginiKQB 7002 Bioinstrumentation Case StudyShafiq JSeahÎncă nu există evaluări
- D-37/1, TTC MIDC, Turbhe, Navi Mumbai-400 703: ThyrocareDocument3 paginiD-37/1, TTC MIDC, Turbhe, Navi Mumbai-400 703: ThyrocareSahil VaishyaÎncă nu există evaluări
- Critical Factors For Successful Real-Time PCRDocument64 paginiCritical Factors For Successful Real-Time PCR7lightbourn5893100% (1)
- PMDC MDCAT Curriculum 2023 Final PDFDocument11 paginiPMDC MDCAT Curriculum 2023 Final PDFmudassar ghouriÎncă nu există evaluări
- Annex 1 COVID 19 NAT Listed TestsDocument35 paginiAnnex 1 COVID 19 NAT Listed TestsMohamed Abdel KareemÎncă nu există evaluări