S-ar putea să vă placă și
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (73)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- PTA014 YMCA Sports Massage ManualDocument58 paginiPTA014 YMCA Sports Massage ManualWilbur AlmedaÎncă nu există evaluări
- Geriatric Medicine - Lally, Frank, Roffe, Christine (SRG)Document237 paginiGeriatric Medicine - Lally, Frank, Roffe, Christine (SRG)pperutti100% (3)
- Chapter 34 Reviewer From Katzung's Basic & Clinical PharmacologyDocument6 paginiChapter 34 Reviewer From Katzung's Basic & Clinical PharmacologyAimee Redor100% (1)
- Perioperative Assessment in Elderly Surgical Patients: Geriatrics Point of ViewDocument43 paginiPerioperative Assessment in Elderly Surgical Patients: Geriatrics Point of ViewAhsan Tanio DaulayÎncă nu există evaluări
- 879 FullDocument7 pagini879 FullAhsan Tanio DaulayÎncă nu există evaluări
- Clinical Implication of Ageing Process and CGA - CZHDocument71 paginiClinical Implication of Ageing Process and CGA - CZHAhsan Tanio DaulayÎncă nu există evaluări
- GeriatriDocument10 paginiGeriatriAhsan Tanio DaulayÎncă nu există evaluări
- Paper Obesity Is The Major Determinant of Elevated C-Reactive Protein in Subjects With The Metabolic SyndromeDocument6 paginiPaper Obesity Is The Major Determinant of Elevated C-Reactive Protein in Subjects With The Metabolic SyndromeAhsan Tanio DaulayÎncă nu există evaluări
- Antimicrobial Treatment Guidelines For Common Infections: June 2016Document56 paginiAntimicrobial Treatment Guidelines For Common Infections: June 2016mario x.p.de araujoÎncă nu există evaluări
- ICD-10 Trg. Tool User - GuideDocument15 paginiICD-10 Trg. Tool User - Guidecckpo100% (1)
- Adult Cardiac Life SupportDocument50 paginiAdult Cardiac Life SupportAhsan Tanio DaulayÎncă nu există evaluări
- ICD-10 Chapter I - Certain Infectious and Parasitic Diseases - WikipediaDocument84 paginiICD-10 Chapter I - Certain Infectious and Parasitic Diseases - WikipediaAhsan Tanio DaulayÎncă nu există evaluări
- Check Your English Vocabulary For MedicineDocument65 paginiCheck Your English Vocabulary For MedicineOlga Poleszak100% (1)
- ICD10Document4 paginiICD10Fern PeñarandaÎncă nu există evaluări
- Adult Cardiac Life SupportDocument50 paginiAdult Cardiac Life SupportAhsan Tanio DaulayÎncă nu există evaluări
- Icd10cm Tabular Addenda 2017Document160 paginiIcd10cm Tabular Addenda 2017Ahsan Tanio DaulayÎncă nu există evaluări
- Battlefield AcupunctureDocument7 paginiBattlefield AcupunctureAhsan Tanio DaulayÎncă nu există evaluări
- Jadwal Jaga Azzakiah Jan 2013, Coba 1Document39 paginiJadwal Jaga Azzakiah Jan 2013, Coba 1Ahsan Tanio DaulayÎncă nu există evaluări
- Effect of Dialysis On Bleeding Time in Chronic Renal FailureDocument5 paginiEffect of Dialysis On Bleeding Time in Chronic Renal FailureAhsan Tanio DaulayÎncă nu există evaluări
- Ijhoscr 7 034Document6 paginiIjhoscr 7 034Ahsan Tanio DaulayÎncă nu există evaluări
- Effect of Dialysis On Bleeding Time in Chronic Renal FailureDocument5 paginiEffect of Dialysis On Bleeding Time in Chronic Renal FailureAhsan Tanio DaulayÎncă nu există evaluări
- List of Antibiotics - Wikipedia, The Free EncyclopediaDocument13 paginiList of Antibiotics - Wikipedia, The Free EncyclopediaAhsan Tanio DaulayÎncă nu există evaluări
- Thrombotic Thrombocytopenic PurpuraDocument5 paginiThrombotic Thrombocytopenic PurpuraAhsan Tanio DaulayÎncă nu există evaluări
- Drug Induce ATnDocument33 paginiDrug Induce ATnAhsan Tanio DaulayÎncă nu există evaluări
- Jurnal 2 - AlbuminuriaDocument6 paginiJurnal 2 - AlbuminuriaAhsan Tanio DaulayÎncă nu există evaluări
- Factor VIIDocument8 paginiFactor VIIAhsan Tanio DaulayÎncă nu există evaluări
- Factor VIIDocument8 paginiFactor VIIAhsan Tanio DaulayÎncă nu există evaluări
- Unconjugated BilirubinDocument9 paginiUnconjugated BilirubinAhsan Tanio DaulayÎncă nu există evaluări
- Factor XDocument6 paginiFactor XAhsan Tanio DaulayÎncă nu există evaluări
- Factor IIDocument7 paginiFactor IIAhsan Tanio DaulayÎncă nu există evaluări
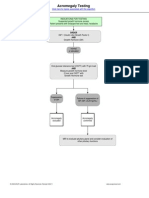
- Acromegaly Testing: Click Here For Topics Associated With This AlgorithmDocument1 paginăAcromegaly Testing: Click Here For Topics Associated With This AlgorithmAhsan Tanio DaulayÎncă nu există evaluări
- Pathology Review Flash CardsDocument137 paginiPathology Review Flash CardsBabak Barghy100% (1)
- Microcatheter Guidewire BrochureDocument8 paginiMicrocatheter Guidewire Brochuresicohod514Încă nu există evaluări
- HEMMORRHOIDECTOMYDocument16 paginiHEMMORRHOIDECTOMYLily CentenoÎncă nu există evaluări
- Pulmonary EmbolismDocument4 paginiPulmonary Embolismemmag1221100% (1)
- Revsed IVDocument123 paginiRevsed IVZhiela Esteban AbivaÎncă nu există evaluări
- Pulmonary EmbolismDocument96 paginiPulmonary Embolismsamice5100% (1)
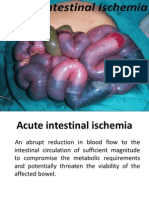
- Acute Intestinal IschemiaDocument50 paginiAcute Intestinal IschemiaRohit ParyaniÎncă nu există evaluări
- Surgical Care Improvement Project JCIDocument50 paginiSurgical Care Improvement Project JCIKania FitrianiÎncă nu există evaluări
- Systematic Review On Risk Factors Related To Central Venous Catheter InfectionDocument18 paginiSystematic Review On Risk Factors Related To Central Venous Catheter InfectionInternational Journal of Innovative Science and Research TechnologyÎncă nu există evaluări
- Anorectal DisorderDocument41 paginiAnorectal DisorderMuhammadÎncă nu există evaluări
- Deep Venous Thrombosis: Anne M. Aquila, APRNDocument20 paginiDeep Venous Thrombosis: Anne M. Aquila, APRNTan RøbìñÎncă nu există evaluări
- Drugs Used in Haematology : Anticoagulants, Antiplatelet Agents and Thrombolytic AgentsDocument42 paginiDrugs Used in Haematology : Anticoagulants, Antiplatelet Agents and Thrombolytic AgentsIrum RafeeqÎncă nu există evaluări
- Rheumatological Disorders in PregnancyDocument61 paginiRheumatological Disorders in Pregnancydrsyan hamzahÎncă nu există evaluări
- Chest: Perioperative Management of Antithrombotic TherapyDocument25 paginiChest: Perioperative Management of Antithrombotic TherapyMahtheerÎncă nu există evaluări
- PATHOLOGY HAQs 2nd EdDocument26 paginiPATHOLOGY HAQs 2nd Edcharusharma9831240259Încă nu există evaluări
- 003 Pathology MCQ ACEM Primary CardiovascularDocument5 pagini003 Pathology MCQ ACEM Primary Cardiovascularbmhsh100% (2)
- Overview of Acute Pulmonary Embolism in AdultsDocument18 paginiOverview of Acute Pulmonary Embolism in AdultscrucaioÎncă nu există evaluări
- Physical Therapy Approaches For Wound CareDocument94 paginiPhysical Therapy Approaches For Wound CarewirdhaÎncă nu există evaluări
- Disseminated Intravascular Coagulopathy Dic 1 1Document25 paginiDisseminated Intravascular Coagulopathy Dic 1 1api-394684626Încă nu există evaluări
- Anticoagulation in The Peripheral Arterial DiseaseDocument7 paginiAnticoagulation in The Peripheral Arterial DiseasePedro Alejandro Saldaña VillaseñorÎncă nu există evaluări
- Khanna 2018Document20 paginiKhanna 2018wahid akbarÎncă nu există evaluări
- Dentistry Questions Final 2016 PDFDocument393 paginiDentistry Questions Final 2016 PDFWasiAliMemon0% (1)
- Peripheral Arterial Occlusive DiseaseDocument4 paginiPeripheral Arterial Occlusive Diseasekrisfred14100% (1)
- Epidemiology and Public Health MedicineDocument200 paginiEpidemiology and Public Health MedicineRakesh100% (1)
- Mohamed Abdel Shafy Mohammady Tabl - 7 - Safety of Ticagrelor Post Fibrinolysis in STEMI PatientsDocument60 paginiMohamed Abdel Shafy Mohammady Tabl - 7 - Safety of Ticagrelor Post Fibrinolysis in STEMI PatientsJovita SardanisÎncă nu există evaluări
- Drug StudyDocument2 paginiDrug StudyAjay SupanÎncă nu există evaluări
- Comparing Venous Stenting With Consevative Treatment in Patien With Deep Venous ObstructionDocument8 paginiComparing Venous Stenting With Consevative Treatment in Patien With Deep Venous ObstructionabdulÎncă nu există evaluări
- MCQS MMDocument45 paginiMCQS MMAyesha .Încă nu există evaluări