S-ar putea să vă placă și
- Russian GrammarDocument141 paginiRussian GrammarMichael Sturgeon, Ph.D.100% (55)
- Integration Exercises (With Answer Key)Document22 paginiIntegration Exercises (With Answer Key)Denise CedeñoÎncă nu există evaluări
- App Math 08 CDocument279 paginiApp Math 08 CYunelsy Nápoles AlvarezÎncă nu există evaluări
- App Math 08 CDocument279 paginiApp Math 08 CYunelsy Nápoles AlvarezÎncă nu există evaluări
- App Math 08 CDocument279 paginiApp Math 08 CYunelsy Nápoles AlvarezÎncă nu există evaluări
- App Math 08 CDocument279 paginiApp Math 08 CYunelsy Nápoles AlvarezÎncă nu există evaluări
- Russian Writing CursiveDocument4 paginiRussian Writing CursiveTanner Gaucher100% (3)
- Water Gas ShiftDocument401 paginiWater Gas ShiftTinker BehrÎncă nu există evaluări
- CatalysisDocument329 paginiCatalysisRakesh ReddyÎncă nu există evaluări
- Magnetic Nanoparticle Based CatalystDocument3 paginiMagnetic Nanoparticle Based CatalystIsabel GarcíaÎncă nu există evaluări
- ApplicationDocument2 paginiApplicationayesha rehmanÎncă nu există evaluări
- New Trends in Design For Sustainability in Modern Industry Through Green ChemistryDocument6 paginiNew Trends in Design For Sustainability in Modern Industry Through Green ChemistryAlexS FigueroaÎncă nu există evaluări
- Rethinking Amide Bond SynthesisDocument9 paginiRethinking Amide Bond SynthesisPaolo SuatingÎncă nu există evaluări
- Isocyanides in the Synthesis of Nitrogen HeterocyclesDocument31 paginiIsocyanides in the Synthesis of Nitrogen HeterocyclesroxideÎncă nu există evaluări
- Amideation Uding Ball MillDocument8 paginiAmideation Uding Ball MillAdamÎncă nu există evaluări
- Catalytic Amide Activation With Thermally StableDocument26 paginiCatalytic Amide Activation With Thermally StableCristhian P. EcheverríaÎncă nu există evaluări
- Organic Photocatalysis: Carbon Nitride Semiconductors vs. Molecular CatalystsDocument16 paginiOrganic Photocatalysis: Carbon Nitride Semiconductors vs. Molecular CatalystsMuhammad UsamaÎncă nu există evaluări
- Assignment No.3: Asad AliDocument4 paginiAssignment No.3: Asad AliMuhammad UsamaÎncă nu există evaluări
- Anie 202007899Document9 paginiAnie 202007899KuniÎncă nu există evaluări
- Major Advances and Challenges in Heterogeneous Catalysis for Environmental ApplicationsDocument26 paginiMajor Advances and Challenges in Heterogeneous Catalysis for Environmental ApplicationsJuan Lopez HernándezÎncă nu există evaluări
- Jing 2019Document27 paginiJing 2019GeraldiineDuqueSalazarÎncă nu există evaluări
- Radical and Ion-pairing Strategies in Asymmetric OrganocatalysisDe la EverandRadical and Ion-pairing Strategies in Asymmetric OrganocatalysisÎncă nu există evaluări
- Research ProposalDocument7 paginiResearch ProposalIkechukwu Ejim100% (2)
- Organometallic Chemistry and Homogeneous CatalysisDocument28 paginiOrganometallic Chemistry and Homogeneous CatalysisJunny CeronÎncă nu există evaluări
- Asian J Org Chem - 2022 - Naik - Recent Trends in Upgrading of CO2 As A C1 Reactant in N and C Methylation ReactionsDocument26 paginiAsian J Org Chem - 2022 - Naik - Recent Trends in Upgrading of CO2 As A C1 Reactant in N and C Methylation ReactionsShailendraÎncă nu există evaluări
- Organo Cat Alys IsDocument15 paginiOrgano Cat Alys IsrajendickÎncă nu există evaluări
- Biocatalysis - Key To Sustainable Industrial ChemistryDocument12 paginiBiocatalysis - Key To Sustainable Industrial ChemistryAntonio Carlos Flash BatistaÎncă nu există evaluări
- Metal CatalystDocument19 paginiMetal CatalystDinesh Babu MÎncă nu există evaluări
- ChemSusChem - 2016 - Schlager - Bio Electrocatalytic Application of Microorganisms For Carbon Dioxide Reduction To MethaneDocument9 paginiChemSusChem - 2016 - Schlager - Bio Electrocatalytic Application of Microorganisms For Carbon Dioxide Reduction To MethaneliviuÎncă nu există evaluări
- Biotransfrmtns Prepartv Organic Chemistry: The Use of Isolated Enzymes and Whole Cell Systems in SynthesisDe la EverandBiotransfrmtns Prepartv Organic Chemistry: The Use of Isolated Enzymes and Whole Cell Systems in SynthesisÎncă nu există evaluări
- Chem. Rev. 2002, 102, 3543Document36 paginiChem. Rev. 2002, 102, 3543kersitoÎncă nu există evaluări
- Stereselective Reactions Calatyzed by LipasesDocument16 paginiStereselective Reactions Calatyzed by LipasesDANICA SEGUROÎncă nu există evaluări
- Conjugated Shape-Persistent Macrocycles Via Schiff-Base Condensation: New Motifs For Supramolecular ChemistryDocument16 paginiConjugated Shape-Persistent Macrocycles Via Schiff-Base Condensation: New Motifs For Supramolecular ChemistryBendjeffal HaceneÎncă nu există evaluări
- Isoquinoline Alkenylboron ACIE 2023Document9 paginiIsoquinoline Alkenylboron ACIE 2023Mutiva YyÎncă nu există evaluări
- Transition Metal-Catalyzed Conversion of Aldehydes To KetonesDocument20 paginiTransition Metal-Catalyzed Conversion of Aldehydes To KetonesjavasoloÎncă nu există evaluări
- Sustainable Production of Benzylamines From LigninDocument6 paginiSustainable Production of Benzylamines From LigninGRagaÎncă nu există evaluări
- Click FFDocument18 paginiClick FFDanesh AzÎncă nu există evaluări
- Sustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeDe la EverandSustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeÎncă nu există evaluări
- Role of Chemical Reaction Engineering in Sustainable Process DevelopmentDocument131 paginiRole of Chemical Reaction Engineering in Sustainable Process DevelopmentjosephusniÎncă nu există evaluări
- Enhanced Bio-Electrochemical Reduction of Carbon Dioxide by Using Neutral Red As A Redox MediatorDocument10 paginiEnhanced Bio-Electrochemical Reduction of Carbon Dioxide by Using Neutral Red As A Redox MediatorliviuÎncă nu există evaluări
- Friend2017 HeteroDocument5 paginiFriend2017 HeteroOmmi Samuel G SÎncă nu există evaluări
- New Green Synthesis Approaches of Pharmacologically Active Heterocyclic CompoundsDocument7 paginiNew Green Synthesis Approaches of Pharmacologically Active Heterocyclic CompoundsEditor IJTSRDÎncă nu există evaluări
- Accepted Manuscript: Coordination Chemistry ReviewsDocument102 paginiAccepted Manuscript: Coordination Chemistry ReviewsKien Pham TÎncă nu există evaluări
- Heterobifunctional PEGs: Efficient StrategiesDocument4 paginiHeterobifunctional PEGs: Efficient StrategiesFranciscoÎncă nu există evaluări
- Thesis On Oxygen HeterocyclesDocument6 paginiThesis On Oxygen Heterocyclesbk1svxmr100% (2)
- Pursuing Practical Elegance in Chemical Synthesis: Ryoji NoyoriDocument5 paginiPursuing Practical Elegance in Chemical Synthesis: Ryoji NoyoriTran Thanh HaÎncă nu există evaluări
- Short Review On Metallocene Complexes: Synthesis, and Biomedical ApplicationsDocument16 paginiShort Review On Metallocene Complexes: Synthesis, and Biomedical ApplicationsInternational Journal of Innovative Science and Research TechnologyÎncă nu există evaluări
- 2023 Molecules 6362 (Orsy)Document14 pagini2023 Molecules 6362 (Orsy)Lázár LászlóÎncă nu există evaluări
- BK9780854041862 00001Document4 paginiBK9780854041862 00001Chrstina Adel TawfiqÎncă nu există evaluări
- Final Report Amit NayakDocument48 paginiFinal Report Amit NayakAmit NayakÎncă nu există evaluări
- 2 PDFDocument13 pagini2 PDFSalman Raza NaqviÎncă nu există evaluări
- 2021H1470293P - Kishor Chougule - APC AssignementDocument30 pagini2021H1470293P - Kishor Chougule - APC AssignementCHOUGULE KISHOR SURYAKANTÎncă nu există evaluări
- Enzyme immobilization methodsDocument14 paginiEnzyme immobilization methodsPrincely ImmanuelÎncă nu există evaluări
- Organic Synthesis DissertationsDocument5 paginiOrganic Synthesis DissertationshcivczwffÎncă nu există evaluări
- SciencedirectDocument24 paginiSciencedirectAna Sansano PérezÎncă nu există evaluări
- Basics of Organic PhotochemistryDocument9 paginiBasics of Organic PhotochemistrySafana SarwarÎncă nu există evaluări
- Research Papers On Synthesis of Heterocyclic CompoundsDocument6 paginiResearch Papers On Synthesis of Heterocyclic Compoundsfwuhlvgkf100% (1)
- Nano Cellulose and Heavy Metal RemovalDocument15 paginiNano Cellulose and Heavy Metal RemovalDr. Ahmed Abdel-HakimÎncă nu există evaluări
- Angew Chem Int Ed - 2022 - Shimoni - Electrostatic Secondary Sphere Interactions That Facilitate Rapid and SelectiveDocument7 paginiAngew Chem Int Ed - 2022 - Shimoni - Electrostatic Secondary Sphere Interactions That Facilitate Rapid and Selectivepandiaraj1988Încă nu există evaluări
- Angewandte: C H FunctionalizationDocument5 paginiAngewandte: C H Functionalizationtofivag317Încă nu există evaluări
- Uamerica 23 01.014Document14 paginiUamerica 23 01.014Rimy Cruz GambaÎncă nu există evaluări
- Impact Fall06Document8 paginiImpact Fall06Steven RankineÎncă nu există evaluări
- Organoiron 1 PDFDocument10 paginiOrganoiron 1 PDFSherly Marcia DevanaÎncă nu există evaluări
- Modern Strategies in Electroorganic SynthesisDocument35 paginiModern Strategies in Electroorganic SynthesisNickly NickÎncă nu există evaluări
- 4丁基间苯二酚几乎没有细胞毒性Document14 pagini4丁基间苯二酚几乎没有细胞毒性jiying huangÎncă nu există evaluări
- 10 1002@cssc 202000098Document8 pagini10 1002@cssc 202000098juliana perez ordoñezÎncă nu există evaluări
- Progress in Microbiology for Fermentative Hydrogen ProductionDocument42 paginiProgress in Microbiology for Fermentative Hydrogen ProductionAnderson TapullimaÎncă nu există evaluări
- Thermodynamics in the production and purification of methanol from methaneDocument23 paginiThermodynamics in the production and purification of methanol from methaneLucas MarchiniÎncă nu există evaluări
- DFGHJKLDocument102 paginiDFGHJKLLucas MarchiniÎncă nu există evaluări
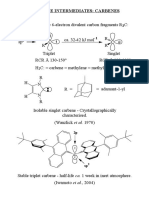
- Reactive Intermediates: CarbenesDocument16 paginiReactive Intermediates: CarbenesvkrÎncă nu există evaluări
- 638 T 01Document116 pagini638 T 01FernyCabezasÎncă nu există evaluări
- Lec 9 Week 12Document36 paginiLec 9 Week 12Lucas MarchiniÎncă nu există evaluări
- ImpropDocument1 paginăImpropLucas MarchiniÎncă nu există evaluări
- Reactive Intermediates: CarbenesDocument16 paginiReactive Intermediates: CarbenesvkrÎncă nu există evaluări
- Strategy and Conclusions: Sum Formula: Calculate The Double-Bond Equivalents From The Sum Formula CDocument3 paginiStrategy and Conclusions: Sum Formula: Calculate The Double-Bond Equivalents From The Sum Formula CLucas MarchiniÎncă nu există evaluări
- Diels AlderDocument8 paginiDiels AlderLucas MarchiniÎncă nu există evaluări
- Sol GelnbvcfghjuDocument22 paginiSol GelnbvcfghjuLucas MarchiniÎncă nu există evaluări
- Capítulo 4 - Técnicas ExperimentalesDocument53 paginiCapítulo 4 - Técnicas Experimentalesleizar_death64Încă nu există evaluări
- Brown Gsas - Harvard 0084L 11009Document198 paginiBrown Gsas - Harvard 0084L 11009Lucas MarchiniÎncă nu există evaluări
- Comparative Studies of Low-Temperature Water-Gas Shift Reaction Over PT Ceo, Au Ceo, and Au Fe O CatalystsDocument7 paginiComparative Studies of Low-Temperature Water-Gas Shift Reaction Over PT Ceo, Au Ceo, and Au Fe O CatalystsLucas MarchiniÎncă nu există evaluări
- Adsorption of Organic Molecules On Rutile TiO2 and Anatase TiO2 Single Crystal SurfacesDocument11 paginiAdsorption of Organic Molecules On Rutile TiO2 and Anatase TiO2 Single Crystal SurfacesLucas MarchiniÎncă nu există evaluări
- c12 Synthesis PDFDocument28 paginic12 Synthesis PDFbencleeseÎncă nu există evaluări
- Sol-Gel Process Chemistry Overview: Solvents, Gelation, Films, Fibers & ApplicationsDocument29 paginiSol-Gel Process Chemistry Overview: Solvents, Gelation, Films, Fibers & ApplicationsLucas MarchiniÎncă nu există evaluări
- Study On The Formation Process of Al2O3-TiO2 Composite PowdersDocument6 paginiStudy On The Formation Process of Al2O3-TiO2 Composite PowdersLucas MarchiniÎncă nu există evaluări
- Effect of Brookite Phase On The Anatase-Rutile Transition in Titania NanoparticlesDocument6 paginiEffect of Brookite Phase On The Anatase-Rutile Transition in Titania NanoparticlesLucas MarchiniÎncă nu există evaluări
- Preparation and Raman Spectrum of Rutile Single Crystals Using Floating Zone MethodDocument3 paginiPreparation and Raman Spectrum of Rutile Single Crystals Using Floating Zone MethodLucas MarchiniÎncă nu există evaluări
- Structure and Properties of TiO2 Surfaces - A Brief ReviewDocument7 paginiStructure and Properties of TiO2 Surfaces - A Brief ReviewLucas MarchiniÎncă nu există evaluări
- A Reviwe of TiO2 NanoparticlesDocument19 paginiA Reviwe of TiO2 NanoparticlesLucas Marchini100% (1)
- Chem. Rev. 115, 9307-9387 (2015) - Organocatalytic Reactions Enabled by NHCsDocument81 paginiChem. Rev. 115, 9307-9387 (2015) - Organocatalytic Reactions Enabled by NHCssunil_vaman_joshiÎncă nu există evaluări
- Umpolung reactivity: methods for interchanging carbonyl donor and acceptor reactivityDocument28 paginiUmpolung reactivity: methods for interchanging carbonyl donor and acceptor reactivitymeauna100% (1)
- Benzoin Condensation ReferencesDocument9 paginiBenzoin Condensation ReferencesGhulam RabbaniÎncă nu există evaluări
- Cs-Mediated Organic TransformationsDocument39 paginiCs-Mediated Organic TransformationsPoonam jamwalÎncă nu există evaluări
- Enantioselective Organocatalysis: Matthew J. Gaunt, Carin C.C. Johansson, Andy Mcnally and Ngoc T. VoDocument20 paginiEnantioselective Organocatalysis: Matthew J. Gaunt, Carin C.C. Johansson, Andy Mcnally and Ngoc T. VoLucas MarchiniÎncă nu există evaluări