S-ar putea să vă placă și
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (120)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (73)
- Nutrition Label Worksheet PDFDocument4 paginiNutrition Label Worksheet PDFCha Marie100% (2)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- CAPE Bio Mark SchemeDocument4 paginiCAPE Bio Mark Schemeron97150% (2)
- Temporal Dynamics of The Human Vaginal MDocument13 paginiTemporal Dynamics of The Human Vaginal MutpalmtbiÎncă nu există evaluări
- Evz 158Document12 paginiEvz 158utpalmtbiÎncă nu există evaluări
- Full PDFDocument6 paginiFull PDFAkarPhyoeKanbauk1Încă nu există evaluări
- Letter: Phylogenetic Evidence For Sino-Tibetan Origin in Northern China in The Late NeolithicDocument14 paginiLetter: Phylogenetic Evidence For Sino-Tibetan Origin in Northern China in The Late NeolithicutpalmtbiÎncă nu există evaluări
- Ncomms 12845Document12 paginiNcomms 12845utpalmtbiÎncă nu există evaluări
- E8822 FullDocument8 paginiE8822 FullutpalmtbiÎncă nu există evaluări
- Other Technical Details of The ProposalDocument2 paginiOther Technical Details of The ProposalutpalmtbiÎncă nu există evaluări
- A Pangenomic Study of Bacillus ThuringiensisDocument10 paginiA Pangenomic Study of Bacillus ThuringiensisutpalmtbiÎncă nu există evaluări
- 15 Things Highly Confident People DoDocument3 pagini15 Things Highly Confident People DoutpalmtbiÎncă nu există evaluări
- Primer DesigningDocument25 paginiPrimer DesigningutpalmtbiÎncă nu există evaluări
- WJTM 3 133Document9 paginiWJTM 3 133utpalmtbiÎncă nu există evaluări
- Raj-Yoga - Swami VivekanandaDocument268 paginiRaj-Yoga - Swami VivekanandautpalmtbiÎncă nu există evaluări
- ReplicationDocument47 paginiReplicationutpalmtbiÎncă nu există evaluări
- Biosynthesis and Functions of Bacillithiol, A Major Low-Molecular-Weight Thiol in BacilliDocument5 paginiBiosynthesis and Functions of Bacillithiol, A Major Low-Molecular-Weight Thiol in BacilliutpalmtbiÎncă nu există evaluări
- What Makes Pathogens PathogenicDocument7 paginiWhat Makes Pathogens PathogenicutpalmtbiÎncă nu există evaluări
- Core Genome Haplotype Diversity and VacA Allelic Heterogeneity of Chinese Helicobacter Pylori StrainsDocument7 paginiCore Genome Haplotype Diversity and VacA Allelic Heterogeneity of Chinese Helicobacter Pylori StrainsutpalmtbiÎncă nu există evaluări
- Analysing Batch Reactor DataDocument3 paginiAnalysing Batch Reactor DatautpalmtbiÎncă nu există evaluări
- Biogeography of The Ecosystems of The Healthy Human BodyDocument56 paginiBiogeography of The Ecosystems of The Healthy Human BodyutpalmtbiÎncă nu există evaluări
- What Is A Gene, Post-EnCODE History and Updated DefinitionDocument14 paginiWhat Is A Gene, Post-EnCODE History and Updated Definitionutpalmtbi100% (1)
- Testing The Infinitely Many Genes Model For The Evolution of The Bacterial Core Genome and PangenomeDocument21 paginiTesting The Infinitely Many Genes Model For The Evolution of The Bacterial Core Genome and PangenomeutpalmtbiÎncă nu există evaluări
- Correlations Between Bacterial Ecology and Mobile DNADocument11 paginiCorrelations Between Bacterial Ecology and Mobile DNAutpalmtbiÎncă nu există evaluări
- A Framework For Classification of Prokaryotic Protein KinasesDocument9 paginiA Framework For Classification of Prokaryotic Protein KinasesutpalmtbiÎncă nu există evaluări
- A Comparison of RpoB and 16S RRNA As Markers in Pyrosequencing Studies of Bacterial DiversityDocument8 paginiA Comparison of RpoB and 16S RRNA As Markers in Pyrosequencing Studies of Bacterial DiversityutpalmtbiÎncă nu există evaluări
- Worldwide Haplotype Diversity and Coding Sequence Variation at Human Bitter Taste Receptor LociDocument6 paginiWorldwide Haplotype Diversity and Coding Sequence Variation at Human Bitter Taste Receptor LociutpalmtbiÎncă nu există evaluări
- The Genetic Structure of Enteric Bacteria From Australian MammalsDocument10 paginiThe Genetic Structure of Enteric Bacteria From Australian MammalsutpalmtbiÎncă nu există evaluări
- Population Structure and Its Effect On Haplotype Diversity and Linkage Disequilibrium Surrounding The Xa5 Locus of Rice (Oryza Sativa L.)Document12 paginiPopulation Structure and Its Effect On Haplotype Diversity and Linkage Disequilibrium Surrounding The Xa5 Locus of Rice (Oryza Sativa L.)utpalmtbiÎncă nu există evaluări
- Statistical Methods For Detecting Molecular AdaptationDocument8 paginiStatistical Methods For Detecting Molecular AdaptationutpalmtbiÎncă nu există evaluări
- Evaluation of Computational Metabolic-Pathway Predictions For Helicobacter PyloriDocument18 paginiEvaluation of Computational Metabolic-Pathway Predictions For Helicobacter PyloriutpalmtbiÎncă nu există evaluări
- Extreme Conservation and Non-Neutral Evolution of The CpmA Circadian Locus in A Globally Distributed Chroococcidiopsis Sp. From Naturally Stressful HabitatsDocument9 paginiExtreme Conservation and Non-Neutral Evolution of The CpmA Circadian Locus in A Globally Distributed Chroococcidiopsis Sp. From Naturally Stressful HabitatsutpalmtbiÎncă nu există evaluări
- Evolutionary Perspective On The Origin of Haitian Cholera Outbreak StrainDocument9 paginiEvolutionary Perspective On The Origin of Haitian Cholera Outbreak StrainutpalmtbiÎncă nu există evaluări
- Chest TubesDocument34 paginiChest TubesMuhd ShafiqÎncă nu există evaluări
- XLD Agar - Manufcture by TM MediaDocument3 paginiXLD Agar - Manufcture by TM MediaKunal VermaÎncă nu există evaluări
- Meriem Chatti - RéférencesDocument22 paginiMeriem Chatti - RéférencesOussama SghaïerÎncă nu există evaluări
- Cell Cycle BrainstormingDocument3 paginiCell Cycle BrainstormingKimberley MendozaÎncă nu există evaluări
- NutritionDocument5 paginiNutritionk,srikanthÎncă nu există evaluări
- EASL 2021 Version 4 NewDocument691 paginiEASL 2021 Version 4 NewGupse Köroğlu AdalıÎncă nu există evaluări
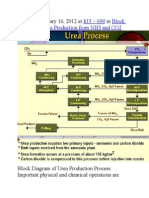
- Published January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2Document9 paginiPublished January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2himanshuchawla654Încă nu există evaluări
- Staar Eoc 2016test Bio F 7Document39 paginiStaar Eoc 2016test Bio F 7api-293216402Încă nu există evaluări
- 3-MS Examples PDFDocument59 pagini3-MS Examples PDFlexuanminhk54lochoaÎncă nu există evaluări
- Pulse Flour From Wheat MillerDocument23 paginiPulse Flour From Wheat MillerCao Trọng HiếuÎncă nu există evaluări
- 8610 - 1 Maham AslamDocument21 pagini8610 - 1 Maham AslamMuhammad BasitÎncă nu există evaluări
- Red Data Book of European Butterflies (Rhopalocera) : Nature and Environment, No. 99Document259 paginiRed Data Book of European Butterflies (Rhopalocera) : Nature and Environment, No. 99TheencyclopediaÎncă nu există evaluări
- AP Biology Practice Exam: Section I: Multiple-Choice QuestionsDocument83 paginiAP Biology Practice Exam: Section I: Multiple-Choice QuestionsKamilla DzhanzakovaÎncă nu există evaluări
- BIOL 3121 - Lecture 13 - 081116Document91 paginiBIOL 3121 - Lecture 13 - 081116Sara HuebnerÎncă nu există evaluări
- Veterinary Gross Anatomy Female Genital TractDocument10 paginiVeterinary Gross Anatomy Female Genital TractTatenda MagejaÎncă nu există evaluări
- Human Genome Editing: Science, Ethics, and GovernanceDocument261 paginiHuman Genome Editing: Science, Ethics, and GovernanceAnonymous 6VKlaeigViÎncă nu există evaluări
- LeaP Science 10 Q4 W3 4Document5 paginiLeaP Science 10 Q4 W3 4D Garcia100% (2)
- Johnson Jerry Alan Chinese Medical Qigong Therapy Vol 5-141-160Document20 paginiJohnson Jerry Alan Chinese Medical Qigong Therapy Vol 5-141-160toanbauÎncă nu există evaluări
- Manage Your Personal Energy PDFDocument46 paginiManage Your Personal Energy PDFezhil manickamÎncă nu există evaluări
- Muscle Biopsy A Diagnostic Tool in Muscle DiseasesDocument9 paginiMuscle Biopsy A Diagnostic Tool in Muscle DiseasesRosa AquinoÎncă nu există evaluări
- The Butterfly Circus Worksheet Activity - 68134Document3 paginiThe Butterfly Circus Worksheet Activity - 68134Hellguz ArtÎncă nu există evaluări
- Accessory Organ of The AbdomenDocument58 paginiAccessory Organ of The AbdomenOgundipe olorunfemiÎncă nu există evaluări
- Jane Goodall BiographyDocument2 paginiJane Goodall BiographyOjas JangalÎncă nu există evaluări
- Recognition, Signaling, and Repair of DNA Double-Strand Breaks Produced by Ionizing Radiation in Mammalian Cells - The Molecular ChoreographyDocument89 paginiRecognition, Signaling, and Repair of DNA Double-Strand Breaks Produced by Ionizing Radiation in Mammalian Cells - The Molecular ChoreographyMaria ClaraÎncă nu există evaluări
- ZIZKA Bromeliaceae ChileDocument22 paginiZIZKA Bromeliaceae ChileJoaquín Eduardo Sepúlveda AstudilloÎncă nu există evaluări
- CV Monica Feliu-Mojer, Ph.D.Document13 paginiCV Monica Feliu-Mojer, Ph.D.moefeliuÎncă nu există evaluări
- Syllabus CompletionDocument1 paginăSyllabus CompletiongopimicroÎncă nu există evaluări
- Principles of Inheritance and Variations: By: Dr. Anand ManiDocument113 paginiPrinciples of Inheritance and Variations: By: Dr. Anand ManiIndu YadavÎncă nu există evaluări