S-ar putea să vă placă și
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- Evidence For DNA As The Hereditary MoleculeDocument2 paginiEvidence For DNA As The Hereditary MoleculeShaezarah MohamudallyÎncă nu există evaluări
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (399)
- Ch10-1 Gen MaterialDocument36 paginiCh10-1 Gen MaterialarcheologistsÎncă nu există evaluări
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- MicropipetteDocument7 paginiMicropipetteMike Serge Razafi0% (1)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (894)
- Ascomycotina Class PPT 2014Document80 paginiAscomycotina Class PPT 2014Shaezarah Mohamudally67% (3)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Gram StainDocument1 paginăGram StainShaezarah MohamudallyÎncă nu există evaluări
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- Malaria PDFDocument36 paginiMalaria PDFYesi Novia AmbaraniÎncă nu există evaluări
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- Identifying Unknown Bacterium-GuideDocument13 paginiIdentifying Unknown Bacterium-GuideShaezarah MohamudallyÎncă nu există evaluări
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Harvard System of ReferenceDocument7 paginiHarvard System of Referencejaya1816Încă nu există evaluări
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (587)
- Biochemical Composition of Fish - A PrimerDocument6 paginiBiochemical Composition of Fish - A PrimerShaezarah MohamudallyÎncă nu există evaluări
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (265)
- Turbidity ScienceDocument26 paginiTurbidity ScienceShaezarah MohamudallyÎncă nu există evaluări
- Similarities Between Prokaryotic and Eukaryotic CellsDocument2 paginiSimilarities Between Prokaryotic and Eukaryotic CellsShaezarah MohamudallyÎncă nu există evaluări
- Malaria Parasites and DiseaseDocument7 paginiMalaria Parasites and DiseaseBalaji Rao NÎncă nu există evaluări
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- Fungus Lec NotesDocument8 paginiFungus Lec NotesShaezarah MohamudallyÎncă nu există evaluări
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (73)
- Characteristics of Fungi N ProtistDocument8 paginiCharacteristics of Fungi N ProtistShaezarah MohamudallyÎncă nu există evaluări
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
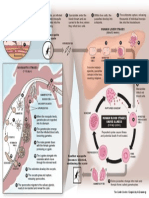
- Malaria Life Cycle ChartDocument1 paginăMalaria Life Cycle ChartShaezarah MohamudallyÎncă nu există evaluări
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- LieDocument1 paginăLieShaezarah MohamudallyÎncă nu există evaluări
- Mal LCDocument4 paginiMal LCreborn007Încă nu există evaluări
- Boltzmann DistributionDocument2 paginiBoltzmann DistributionShaezarah MohamudallyÎncă nu există evaluări
- Lecture of Organic ChemistryDocument13 paginiLecture of Organic ChemistryNutDen R X NuttapongÎncă nu există evaluări
- Growth Curves of Micro-OrganismsDocument31 paginiGrowth Curves of Micro-OrganismsShaezarah MohamudallyÎncă nu există evaluări
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- Topic3 BIOL1030NRDocument11 paginiTopic3 BIOL1030NRShaezarah MohamudallyÎncă nu există evaluări
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2219)
- PolarityDocument1 paginăPolarityDeepakÎncă nu există evaluări
- Fungus Lec NotesDocument8 paginiFungus Lec NotesShaezarah MohamudallyÎncă nu există evaluări
- Microsoft PowerPoint - Bio1009ch13Document48 paginiMicrosoft PowerPoint - Bio1009ch13Shaezarah MohamudallyÎncă nu există evaluări
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- Lecture of Organic ChemistryDocument13 paginiLecture of Organic ChemistryNutDen R X NuttapongÎncă nu există evaluări
- Diff Cent PDFDocument33 paginiDiff Cent PDFShaezarah MohamudallyÎncă nu există evaluări
- Cell Counting Using A HemacytometerDocument3 paginiCell Counting Using A HemacytometerKris Cahyo Mulyatno100% (1)
- RAlaminarflowcabinets PDFDocument4 paginiRAlaminarflowcabinets PDFShaezarah MohamudallyÎncă nu există evaluări
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (119)
- ChemistryDocument9 paginiChemistryShaezarah MohamudallyÎncă nu există evaluări
- CSI Polymerase Chain Reaction Lab ManualDocument6 paginiCSI Polymerase Chain Reaction Lab ManualDank MoviesÎncă nu există evaluări
- Analytical Method PDFDocument85 paginiAnalytical Method PDFiaderzÎncă nu există evaluări
- DNA From Caviar': NCBE, University of ReadingDocument4 paginiDNA From Caviar': NCBE, University of ReadingYosh MidhrasÎncă nu există evaluări
- Colloids and Surfaces A: Physicochemical and Engineering AspectsDocument7 paginiColloids and Surfaces A: Physicochemical and Engineering Aspectsandreea g.Încă nu există evaluări
- A Basic PCR ProtocolDocument4 paginiA Basic PCR ProtocolIrfan HussainÎncă nu există evaluări
- Microbial diversity of indigenous microorganism communities from Vietnam agro-ecosystemsDocument12 paginiMicrobial diversity of indigenous microorganism communities from Vietnam agro-ecosystemsrahmanÎncă nu există evaluări
- Developing Best-Practice Models For The Therapeutic Use of Extracellular VesiclesDocument10 paginiDeveloping Best-Practice Models For The Therapeutic Use of Extracellular VesicleschenÎncă nu există evaluări
- Protein Electrophoresis GEDocument82 paginiProtein Electrophoresis GEIsa Ribeiro100% (1)
- Ape Lex 2015Document106 paginiApe Lex 2015Bolawali KiÎncă nu există evaluări
- 7.biochemical and Structural Characterization of Sturgeon FishDocument10 pagini7.biochemical and Structural Characterization of Sturgeon FishJonathanCoralÎncă nu există evaluări
- Biochem PDFDocument53 paginiBiochem PDFMiriam JonesÎncă nu există evaluări
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- Heat Number in MilkDocument17 paginiHeat Number in MilkLaura MarcelaÎncă nu există evaluări
- Spife TouchDocument4 paginiSpife TouchRita LeongÎncă nu există evaluări
- A Short Review On Proteomics and Its ApplicationsDocument9 paginiA Short Review On Proteomics and Its ApplicationsMySCRIBDÎncă nu există evaluări
- Chapter 11 Biotechnology Principle and ProcessDocument50 paginiChapter 11 Biotechnology Principle and ProcessMkisneymar Meymar100% (1)
- Rapd Profile For Lab ReportDocument4 paginiRapd Profile For Lab Reportapi-341432127Încă nu există evaluări
- Table of ContentsDocument582 paginiTable of Contentsno contractÎncă nu există evaluări
- Western Blot ProtocolDocument6 paginiWestern Blot ProtocollinhÎncă nu există evaluări
- ElectrophoresisDocument38 paginiElectrophoresisHennah Usman67% (3)
- Go TaqDocument2 paginiGo TaqYogi ArismetÎncă nu există evaluări
- USP34-NF29 IndexDocument72 paginiUSP34-NF29 IndexAraya Rattanavichian100% (2)
- Good's Buffers (Biological Buffers) : Products Description / OverviewDocument9 paginiGood's Buffers (Biological Buffers) : Products Description / OverviewCHIRANJEEVIÎncă nu există evaluări
- Statistical Analysis For Plant Tissue Culture DataDocument68 paginiStatistical Analysis For Plant Tissue Culture Datadolt2111Încă nu există evaluări
- For The PA 800 Plus Pharmaceutical Analysis System: SDS-MW Analysis KitDocument42 paginiFor The PA 800 Plus Pharmaceutical Analysis System: SDS-MW Analysis KitThanh Tâm TrầnÎncă nu există evaluări
- Factors Affecting ElectrophoresisDocument20 paginiFactors Affecting ElectrophoresisMuhammad Ramzan0% (1)
- Gel Electrophorosis MpatDocument19 paginiGel Electrophorosis Mpatkavya nainitaÎncă nu există evaluări
- Vikash Kumar: Career ObjectiveDocument2 paginiVikash Kumar: Career ObjectiveAnikesh SinghÎncă nu există evaluări
- Exp 7-SDS-PAGEDocument18 paginiExp 7-SDS-PAGERadwan M SaadehÎncă nu există evaluări
- User Manual: Macherey-NagelDocument34 paginiUser Manual: Macherey-NagelBatuli MwaijandeÎncă nu există evaluări
- 2022 - Biochemistry - Esha D. Dalvie, Jordan C. Stacy, Keir C. Neuman - Recognition of DNA Supercoil Handedness During Catenation Catalyzed byDocument33 pagini2022 - Biochemistry - Esha D. Dalvie, Jordan C. Stacy, Keir C. Neuman - Recognition of DNA Supercoil Handedness During Catenation Catalyzed byjimgogreatÎncă nu există evaluări
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeDe la EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeEvaluare: 4.5 din 5 stele4.5/5 (3)
- Stuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldDe la EverandStuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldEvaluare: 4 din 5 stele4/5 (289)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeDe la EverandChemistry for Breakfast: The Amazing Science of Everyday LifeEvaluare: 4.5 din 5 stele4.5/5 (14)
- Guidelines for Asset Integrity ManagementDe la EverandGuidelines for Asset Integrity ManagementEvaluare: 5 din 5 stele5/5 (1)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction in the Science of Everyday LifeDe la EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction in the Science of Everyday LifeEvaluare: 4 din 5 stele4/5 (9)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsDe la EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsEvaluare: 4 din 5 stele4/5 (146)
- Science Goes Viral: Captivating Accounts of Science in Everyday LifeDe la EverandScience Goes Viral: Captivating Accounts of Science in Everyday LifeEvaluare: 5 din 5 stele5/5 (1)