S-ar putea să vă placă și
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Remediating Motor ControlDocument66 paginiRemediating Motor Controlhis.thunder122100% (3)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- Transcription and Translation Practice WorksheetDocument4 paginiTranscription and Translation Practice Worksheetsmith joeÎncă nu există evaluări
- Duchenne Muscular DystrophyDocument32 paginiDuchenne Muscular DystrophyMr.P.Ramesh, Faculty of Physiotherapy, SRUÎncă nu există evaluări
- Experimental and Quasi-Experimental Designs For Generalized Causal Inference PDFDocument81 paginiExperimental and Quasi-Experimental Designs For Generalized Causal Inference PDFGonçalo SampaioÎncă nu există evaluări
- An Analysis of Mario Castelnuovo-Tedescos Vogelweide - Song CycleDocument95 paginiAn Analysis of Mario Castelnuovo-Tedescos Vogelweide - Song Cycleantonio_forgione100% (2)
- M (ZD (Cx-5: Smart Start GuideDocument42 paginiM (ZD (Cx-5: Smart Start Guideantonio_forgioneÎncă nu există evaluări
- Semi-Detailed Lesson Plan For Cellular RespirationDocument2 paginiSemi-Detailed Lesson Plan For Cellular RespirationZaifel Pacillos100% (5)
- AGU Friday Daily NewspaperDocument56 paginiAGU Friday Daily Newspaperantonio_forgioneÎncă nu există evaluări
- 2013 Fruit Share Is Sold OUTDocument2 pagini2013 Fruit Share Is Sold OUTantonio_forgioneÎncă nu există evaluări
- EddyPro5 User GuideDocument297 paginiEddyPro5 User Guideantonio_forgione100% (1)
- Egu2011 9984Document1 paginăEgu2011 9984antonio_forgioneÎncă nu există evaluări
- Mathed 1Document9 paginiMathed 1antonio_forgioneÎncă nu există evaluări
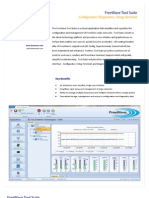
- Tool Suite Overview 11.9.11Document2 paginiTool Suite Overview 11.9.11antonio_forgioneÎncă nu există evaluări
- Egu2010 13937Document1 paginăEgu2010 13937antonio_forgioneÎncă nu există evaluări
- Unisonic Technologies Co.,: Low Voltage Audio Power AmplifierDocument7 paginiUnisonic Technologies Co.,: Low Voltage Audio Power Amplifierantonio_forgioneÎncă nu există evaluări
- Pump in Style Battery Pack Instructions: Important SafeguardsDocument2 paginiPump in Style Battery Pack Instructions: Important Safeguardsantonio_forgioneÎncă nu există evaluări
- Vs10Xx Appnote: Connecting Analog OutputsDocument10 paginiVs10Xx Appnote: Connecting Analog Outputsantonio_forgioneÎncă nu există evaluări
- The Why UI: Using Goal Networks To Improve User InterfacesDocument4 paginiThe Why UI: Using Goal Networks To Improve User Interfacesantonio_forgioneÎncă nu există evaluări
- IMSLP13264-Hummel - Pot-Pourri For Guitar and Piano Op.53Document13 paginiIMSLP13264-Hummel - Pot-Pourri For Guitar and Piano Op.53antonio_forgioneÎncă nu există evaluări
- 2013 ScheduleDocument2 pagini2013 Scheduleantonio_forgioneÎncă nu există evaluări
- Moonlight On The Ocean Cherry, J. W. Weiland, Francis Carpenter, J. E. Notated Music Songs With GuitarDocument5 paginiMoonlight On The Ocean Cherry, J. W. Weiland, Francis Carpenter, J. E. Notated Music Songs With Guitarantonio_forgioneÎncă nu există evaluări
- Immunoproteasome: Dania Mohammad Abu NawasDocument27 paginiImmunoproteasome: Dania Mohammad Abu NawasDania MohÎncă nu există evaluări
- XI NEET - Dakshana Test ScheduleDocument5 paginiXI NEET - Dakshana Test SchedulePoornima K TÎncă nu există evaluări
- Hellagrolip Nitrogenous Brochure enDocument2 paginiHellagrolip Nitrogenous Brochure enBashir A. SialÎncă nu există evaluări
- Paleoecology Concepts and Applications PDFDocument2 paginiPaleoecology Concepts and Applications PDFLaura AvilaÎncă nu există evaluări
- Genetic Disorders in Arab Populations: Qatar: Tawfeg Ben-Omran, Atqah Abdul WahabDocument6 paginiGenetic Disorders in Arab Populations: Qatar: Tawfeg Ben-Omran, Atqah Abdul WahabUmair ZubairÎncă nu există evaluări
- 1,4 ButanediolDocument6 pagini1,4 ButanediolVictor VikeneÎncă nu există evaluări
- Understanding: Bacteria:: Teacher's GuideDocument8 paginiUnderstanding: Bacteria:: Teacher's GuideKari Kristine Hoskins BarreraÎncă nu există evaluări
- Water Quality ManagementDocument61 paginiWater Quality ManagementRahul RanjanÎncă nu există evaluări
- Craetagusarticle PDFDocument7 paginiCraetagusarticle PDFKatty AcostaÎncă nu există evaluări
- Biochemistry of GlycoproteinDocument6 paginiBiochemistry of GlycoproteinMahathir Mohmed100% (7)
- Comparative Analysis of Agrobiological Traits of Durum (T. Durum Desf.) and Bread Wheat (T. Aestivum L.) Varieties in The Karabakh RegionDocument9 paginiComparative Analysis of Agrobiological Traits of Durum (T. Durum Desf.) and Bread Wheat (T. Aestivum L.) Varieties in The Karabakh RegionPublisher NasirÎncă nu există evaluări
- El KoalaDocument4 paginiEl KoalaGerzain OrozcoÎncă nu există evaluări
- Fossils QuizDocument2 paginiFossils Quizapi-322659329Încă nu există evaluări
- Sale Catalog - Diamond Genetics Online Elite Bull SaleDocument48 paginiSale Catalog - Diamond Genetics Online Elite Bull SaleHolstein PlazaÎncă nu există evaluări
- Hope Jennings - Dystopias of Matriarchal Power: Deconstructing The Womb in Angela Carter's Heroes and Villains and The Passion of New EveDocument11 paginiHope Jennings - Dystopias of Matriarchal Power: Deconstructing The Womb in Angela Carter's Heroes and Villains and The Passion of New Evedanutza123Încă nu există evaluări
- Chelex 100-Instruction ManualDocument14 paginiChelex 100-Instruction Manualu77Încă nu există evaluări
- Substance Id enDocument118 paginiSubstance Id ensmaps001Încă nu există evaluări
- Cointegrated VectorsDocument2 paginiCointegrated Vectorsdevikamurugan124206Încă nu există evaluări
- Week 4 SimDocument3 paginiWeek 4 SimJEZUE REMULTAÎncă nu există evaluări
- Estudio Etnobotanico MonzanbiqueDocument11 paginiEstudio Etnobotanico MonzanbiqueJavier JalilieÎncă nu există evaluări
- Slotwise Details July-November 2021Document138 paginiSlotwise Details July-November 2021Ch PrasadÎncă nu există evaluări
- Furtado 2016Document10 paginiFurtado 2016zzzzÎncă nu există evaluări
- R.S 10 MLDocument13 paginiR.S 10 MLShofwatul nadiaÎncă nu există evaluări
- Separation of SubstancesDocument4 paginiSeparation of SubstancessangopsÎncă nu există evaluări
- Bio 310Document39 paginiBio 310flipside1o1Încă nu există evaluări