S-ar putea să vă placă și
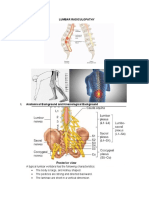
- Lumbar Spine Surgery Neurological ComplicationsDocument9 paginiLumbar Spine Surgery Neurological ComplicationsNURULÎncă nu există evaluări
- RTC Strengthening Article PDFDocument14 paginiRTC Strengthening Article PDFineedsheetzÎncă nu există evaluări
- 01 Pathophysiology of PainDocument3 pagini01 Pathophysiology of PainBani ZakiyahÎncă nu există evaluări
- Learning and Cognitive Flexibility - Frontostriatal Function and Monoaminergic ModulationDocument6 paginiLearning and Cognitive Flexibility - Frontostriatal Function and Monoaminergic ModulationFrancisco Ahumada MéndezÎncă nu există evaluări
- Class II Malocclusion CamouflageDocument48 paginiClass II Malocclusion CamouflageAndreea-Georgiana BălanÎncă nu există evaluări
- A Framework For Exercise Prescription: DOI: 10.1097/TGR.0000000000000011Document23 paginiA Framework For Exercise Prescription: DOI: 10.1097/TGR.0000000000000011Lft Ediel PiñaÎncă nu există evaluări
- Central Sensitisation Another Label or Useful DiagnosisDocument4 paginiCentral Sensitisation Another Label or Useful DiagnosisMohamed ElMeligieÎncă nu există evaluări
- Inflammatory Muscle Diseases NEJM 2015 Marinos DalakasDocument14 paginiInflammatory Muscle Diseases NEJM 2015 Marinos DalakasWalther Seven VGÎncă nu există evaluări
- Caso NEJM 27-2017Document9 paginiCaso NEJM 27-2017Leonardo PgÎncă nu există evaluări
- PruritusDocument10 paginiPruritusdikka ayuniÎncă nu există evaluări
- Mike ReinoldDocument20 paginiMike ReinoldVikas Kashnia0% (1)
- Practical Approach To Common Electrolyte EmergenciesDocument64 paginiPractical Approach To Common Electrolyte EmergenciesSamer MatarÎncă nu există evaluări
- Necessary Elements of A Dermatologic History and Physical Evaluation PDFDocument9 paginiNecessary Elements of A Dermatologic History and Physical Evaluation PDFkyle31Încă nu există evaluări
- Necroptosis PDFDocument11 paginiNecroptosis PDFdavid zambranoÎncă nu există evaluări
- 4.the Pathogenesis of Psoriatic ArthritisDocument12 pagini4.the Pathogenesis of Psoriatic ArthritisstuckinbedÎncă nu există evaluări
- Staged Approach For Rehabilitation ClassificationDocument10 paginiStaged Approach For Rehabilitation ClassificationCambriaChicoÎncă nu există evaluări
- Acido Base Stewart NEJM 2014Document11 paginiAcido Base Stewart NEJM 2014Aracely VIÎncă nu există evaluări
- Anterior Cruciate Ligament Tear NEJMDocument8 paginiAnterior Cruciate Ligament Tear NEJMRui ViegasÎncă nu există evaluări
- Neck PainDocument1 paginăNeck PainHasan RahmanÎncă nu există evaluări
- MSSE-Central Peripheral Fatigue InteractionDocument7 paginiMSSE-Central Peripheral Fatigue InteractionMari PaoÎncă nu există evaluări
- Hypoxia and InflammationDocument10 paginiHypoxia and InflammationAbel Espinoza MedallaÎncă nu există evaluări
- Acute Pain: Mechanisms, Management, and Treatment OptionsDocument61 paginiAcute Pain: Mechanisms, Management, and Treatment OptionsAnnisa RamadhanyÎncă nu există evaluări
- Carpal Tunnel Syndrome - Clinical Manifestations and Diagnosis - UpToDateDocument15 paginiCarpal Tunnel Syndrome - Clinical Manifestations and Diagnosis - UpToDateFajar HerbowoÎncă nu există evaluări
- Treatment of Tendinopathy What Works, What Does Not, and What Is On The Horizon PDFDocument16 paginiTreatment of Tendinopathy What Works, What Does Not, and What Is On The Horizon PDFFranciscoJavierRoblesÎncă nu există evaluări
- Amyotrophic Lateral SclerosisDocument3 paginiAmyotrophic Lateral SclerosisdrusmansaleemÎncă nu există evaluări
- Reinold - Glenohumeral and Scapulothoracic RehabDocument45 paginiReinold - Glenohumeral and Scapulothoracic Rehabshivnair100% (2)
- Measureable Changes in The Neuro-Endocrinal Mechanism Following Spinal Manipulation - 2015Document6 paginiMeasureable Changes in The Neuro-Endocrinal Mechanism Following Spinal Manipulation - 2015Renan O. Pravatta PivettaÎncă nu există evaluări
- 231a - Reconsidering The Way We Look at Movement-VCUDocument19 pagini231a - Reconsidering The Way We Look at Movement-VCUwolfgate0% (1)
- The NTS-PGi-LC-CA1 pathway is important for object recognition memory consolidationDocument8 paginiThe NTS-PGi-LC-CA1 pathway is important for object recognition memory consolidationRehaan FayazÎncă nu există evaluări
- Parkinson's DiseaseDocument17 paginiParkinson's DiseaseRafael Carrillo-BaylonÎncă nu există evaluări
- Research Report: Effect of Neck Exercise On Sitting Posture in Patients With Chronic Neck PainDocument10 paginiResearch Report: Effect of Neck Exercise On Sitting Posture in Patients With Chronic Neck Painapi-2368197Încă nu există evaluări
- Management of Acute Hip Fracture - NEJM PDFDocument10 paginiManagement of Acute Hip Fracture - NEJM PDFRui ViegasÎncă nu există evaluări
- Lumbar Facet Syndrome - BogdukDocument8 paginiLumbar Facet Syndrome - BogdukSamSchroetkeÎncă nu există evaluări
- Mast Cells, Glia and Neuroinflammation Partners in CrimeDocument14 paginiMast Cells, Glia and Neuroinflammation Partners in CrimeCelia Steiman100% (1)
- Vestibular System PrimerDocument17 paginiVestibular System PrimerfadhillaÎncă nu există evaluări
- Evidence-Based Examination and Intervention of the Hip JointDocument10 paginiEvidence-Based Examination and Intervention of the Hip JointRicha GahuniaÎncă nu există evaluări
- Hand Fractures PDFDocument92 paginiHand Fractures PDFfiestaÎncă nu există evaluări
- Thoracic Outlet Syndrome - Shoulder & Elbow - OrthobulletsDocument13 paginiThoracic Outlet Syndrome - Shoulder & Elbow - OrthobulletsSylvia GraceÎncă nu există evaluări
- Monaco Treatment Planning Enhances Departmental EfficienciesDocument9 paginiMonaco Treatment Planning Enhances Departmental Efficienciesricky rdnÎncă nu există evaluări
- Urogenital Symptoms in Neurologic Patients.14Document20 paginiUrogenital Symptoms in Neurologic Patients.14Fawad JanÎncă nu există evaluări
- Acupuncture Points and Multireceptive NeuronsDocument9 paginiAcupuncture Points and Multireceptive NeuronsmtamilmaniÎncă nu există evaluări
- An Adherent Nerve Root - Classification and Exercise Therapy in A PatientDocument4 paginiAn Adherent Nerve Root - Classification and Exercise Therapy in A PatientPablo Zamora MillanÎncă nu există evaluări
- Allen 2017Document22 paginiAllen 2017Jere AyerbeÎncă nu există evaluări
- Lumbar Radiculopathy Medback Castillo Mendez EDITEDDocument12 paginiLumbar Radiculopathy Medback Castillo Mendez EDITEDSteve ColbertÎncă nu există evaluări
- The Effect of Therapeutic Ultrasound On Metallic Implants - A Study in RatsDocument5 paginiThe Effect of Therapeutic Ultrasound On Metallic Implants - A Study in RatsnatkwqÎncă nu există evaluări
- Osteoarthritis of The KneeDocument8 paginiOsteoarthritis of The Kneeas3syamut649250% (2)
- Bartold S.J. (2004) The Plantar Fascia As Souce of PainDocument13 paginiBartold S.J. (2004) The Plantar Fascia As Souce of PainManuel DamilÎncă nu există evaluări
- Mechanisms of Cardiac Pain PDFDocument27 paginiMechanisms of Cardiac Pain PDFLa Torre LeoÎncă nu există evaluări
- Kaltenborn 1993Document5 paginiKaltenborn 1993hari vijayÎncă nu există evaluări
- Spinal Biomechanics ShortDocument10 paginiSpinal Biomechanics ShortkkdÎncă nu există evaluări
- 2023 AAN Myositis Autoantibodies FINALDocument53 pagini2023 AAN Myositis Autoantibodies FINALEvelina ȘabanovÎncă nu există evaluări
- Neurogenic BowlDocument21 paginiNeurogenic BowlFaridatul IsniyahÎncă nu există evaluări
- G24 Fragility FracturesDocument38 paginiG24 Fragility FracturesDeep Katyan DeepÎncă nu există evaluări
- AnkleDocument54 paginiAnkleUmi ShakiraÎncă nu există evaluări
- Lumbar Radicular Pain: PathophysiologyDocument4 paginiLumbar Radicular Pain: PathophysiologynetifarhatiiÎncă nu există evaluări
- Tendon Neuroplastic TrainingDocument8 paginiTendon Neuroplastic TrainingFrantzesco KangarisÎncă nu există evaluări
- Multiple-System AtrophyDocument15 paginiMultiple-System AtrophyNathasia SuryawijayaÎncă nu există evaluări
- Multiple System Atrophy: Cellular and Molecular Pathology: D J Burn, E JarosDocument8 paginiMultiple System Atrophy: Cellular and Molecular Pathology: D J Burn, E Jarosuzair khanÎncă nu există evaluări
- Frontotemporal Dementia NeuropathologyDocument14 paginiFrontotemporal Dementia Neuropathologyppico1963Încă nu există evaluări
- Alpha-Synuclein Secretion and DysfunctionDocument7 paginiAlpha-Synuclein Secretion and Dysfunctionbenghoe77Încă nu există evaluări
- Myoclonus With DementiaDocument8 paginiMyoclonus With DementiaSantosh DashÎncă nu există evaluări
- Msa Review ArticleDocument15 paginiMsa Review ArticleSantosh DashÎncă nu există evaluări
- Review Article: Tuberculous Meningitis: Diagnosis and Treatment OverviewDocument10 paginiReview Article: Tuberculous Meningitis: Diagnosis and Treatment OverviewMeldaÎncă nu există evaluări
- Emg BasicsDocument5 paginiEmg BasicsSantosh DashÎncă nu există evaluări
- Cerebellum in CognitionDocument24 paginiCerebellum in CognitionSantosh DashÎncă nu există evaluări
- 2 Blood Brain BarrierDocument13 pagini2 Blood Brain BarrierEva FonsecaÎncă nu există evaluări
- Spotters 1Document10 paginiSpotters 1elavarkuzhali2019Încă nu există evaluări
- Abnormal Psychology TestDocument7 paginiAbnormal Psychology TestCamae Snyder GangcuangcoÎncă nu există evaluări
- Product Name:: Alaris™ GS, GH, CC, TIVA, PK, Enteral Syringe PumpDocument14 paginiProduct Name:: Alaris™ GS, GH, CC, TIVA, PK, Enteral Syringe PumpSalim AloneÎncă nu există evaluări
- Short-Term Clinical Outcomes and Safety Associated With Percutaneous Radiofrequency Treatment For Excessive SweatingDocument10 paginiShort-Term Clinical Outcomes and Safety Associated With Percutaneous Radiofrequency Treatment For Excessive SweatingsadraÎncă nu există evaluări
- The Expanded Program On ImmunizationDocument26 paginiThe Expanded Program On ImmunizationJudee Marie MalubayÎncă nu există evaluări
- Prescription AnalysisDocument16 paginiPrescription AnalysisMohd Azfar HafizÎncă nu există evaluări
- Science 2013 Couzin Frankel 68 9Document2 paginiScience 2013 Couzin Frankel 68 9Ricardo ChavarriaÎncă nu există evaluări
- Question 1Document25 paginiQuestion 1Anonymous 1T0qSzPt1PÎncă nu există evaluări
- Cipp 2015 AbstractsDocument88 paginiCipp 2015 AbstractsBuyanaaRise100% (2)
- The Geriatric Anxiety Inventory in Primary CareDocument4 paginiThe Geriatric Anxiety Inventory in Primary CareYudistiro Adi NugrohoÎncă nu există evaluări
- Certification of Health Care Provider For Family MBRDocument4 paginiCertification of Health Care Provider For Family MBRapi-241312847Încă nu există evaluări
- Pricelist 13 Juli 2020Document21 paginiPricelist 13 Juli 2020Achmad Sya'idÎncă nu există evaluări
- 009the Automated Nucleated Red Cell Count On The Sysmex XE-2100Document5 pagini009the Automated Nucleated Red Cell Count On The Sysmex XE-2100blanket_thÎncă nu există evaluări
- Stress-Related Mucosal Disease: Mitchell J. Spirt, MDDocument11 paginiStress-Related Mucosal Disease: Mitchell J. Spirt, MDnur hidayatÎncă nu există evaluări
- Medical Surgical Nursing Practice Test Part 1Document15 paginiMedical Surgical Nursing Practice Test Part 1cleznielÎncă nu există evaluări
- Fraktur DentoalveolarDocument25 paginiFraktur DentoalveolarfirmansyahddsÎncă nu există evaluări
- Acquired Brain Injury Early Rehabilitation and Long Term OutcomeDocument46 paginiAcquired Brain Injury Early Rehabilitation and Long Term OutcomeVeena RaigangarÎncă nu există evaluări
- DURVA - (Harali - Jeevani) Cynodon Dactylon (An Ordinary Grass With Miracles)Document4 paginiDURVA - (Harali - Jeevani) Cynodon Dactylon (An Ordinary Grass With Miracles)vijaysai77100% (1)
- Registered Pharmacist Affidavit FormatDocument1 paginăRegistered Pharmacist Affidavit FormatSyed NawazÎncă nu există evaluări
- I. The Problem and Its Background Ii. Demand and Analysis Review of Related Literature and Case StudiesDocument3 paginiI. The Problem and Its Background Ii. Demand and Analysis Review of Related Literature and Case StudiesPaulyn Mae Dela CruzÎncă nu există evaluări
- Heart Rate LabDocument6 paginiHeart Rate LabSteve RodriguesÎncă nu există evaluări
- Wojejuterer Health Assessment in Nursing 5th Edition PupegDocument3 paginiWojejuterer Health Assessment in Nursing 5th Edition Pupegace Decla100% (1)
- Designer No Auxilio Ao Cobate Da Covid 19Document1 paginăDesigner No Auxilio Ao Cobate Da Covid 19suzy luaÎncă nu există evaluări
- The Federation of Motor Sports Clubs of India 2022 Appendix "B" - Medical HistoryDocument2 paginiThe Federation of Motor Sports Clubs of India 2022 Appendix "B" - Medical HistoryMBG No.14Încă nu există evaluări
- Nama: Stevany Ayuningsi Aduga Nim: 150600 Kelas: Keperawatan B Questioning To Fill in Pain Assessment Form ObjectivesDocument8 paginiNama: Stevany Ayuningsi Aduga Nim: 150600 Kelas: Keperawatan B Questioning To Fill in Pain Assessment Form ObjectivesAnathasya SalamatÎncă nu există evaluări
- Conservative Management of Cutaneous Sinus Tract of Dental Origin Report of Two CasesDocument4 paginiConservative Management of Cutaneous Sinus Tract of Dental Origin Report of Two CasesInternational Journal of Innovative Science and Research TechnologyÎncă nu există evaluări
- Greeks Were One of The First Patrons of This ProfessionDocument1 paginăGreeks Were One of The First Patrons of This ProfessionpradipdeshmukhÎncă nu există evaluări
- Konsep Pembangunan Kesehatan Di Indonesia-1Document8 paginiKonsep Pembangunan Kesehatan Di Indonesia-1PIPITÎncă nu există evaluări
- GE - MS Accessories GuideDocument50 paginiGE - MS Accessories GuidemaruthaiÎncă nu există evaluări
- BPH Zinc GreenTea Papaya LeafDocument13 paginiBPH Zinc GreenTea Papaya Leafteddy_shashaÎncă nu există evaluări