S-ar putea să vă placă și
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (74)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- Method Statement of Pipeline WorksDocument13 paginiMethod Statement of Pipeline Worksमनिसभेटुवाल86% (21)
- Carpentry: Exploratory CourseDocument25 paginiCarpentry: Exploratory CourseJohn Nelson Picones100% (3)
- Light Object DSP Controller Setup GuideDocument20 paginiLight Object DSP Controller Setup Guideblondu0070% (1)
- Chrony Beta and GammaDocument36 paginiChrony Beta and GammaÁdám MajorÎncă nu există evaluări
- Lab 2.1Document2 paginiLab 2.1Manjari Srivastava100% (3)
- Geomatric Nonlinearity Formulation in OpenseesDocument16 paginiGeomatric Nonlinearity Formulation in Openseessami_bangash_1Încă nu există evaluări
- Aggregate Durability Physical PropertiesDocument32 paginiAggregate Durability Physical Propertiessami_bangash_1Încă nu există evaluări
- Static and Dynamic Analysis of High Rise BuildingsDocument13 paginiStatic and Dynamic Analysis of High Rise Buildingssami_bangash_1100% (1)
- CEE 572: Earthquake Engineering Assignment 2 Spring 2017Document1 paginăCEE 572: Earthquake Engineering Assignment 2 Spring 2017sami_bangash_1Încă nu există evaluări
- 2000 T.R. Higgins Award Paper - A Practical Look at Frame Analysis, Stability and Leaning ColumnsDocument15 pagini2000 T.R. Higgins Award Paper - A Practical Look at Frame Analysis, Stability and Leaning ColumnsSamuel PintoÎncă nu există evaluări
- Seismic Risk To Transportation NetworksDocument1 paginăSeismic Risk To Transportation Networkssami_bangash_1Încă nu există evaluări
- Elastic Column Buckling and The Effect of End RestraintDocument2 paginiElastic Column Buckling and The Effect of End RestraintSandeep VaishnavÎncă nu există evaluări
- Masonry Mortar Strength OverviewDocument2 paginiMasonry Mortar Strength Overviewsami_bangash_1Încă nu există evaluări
- Soil Mechanics Uiuc SyllabusDocument3 paginiSoil Mechanics Uiuc Syllabussami_bangash_1Încă nu există evaluări
- Math415 Lec4Document8 paginiMath415 Lec4sami_bangash_1Încă nu există evaluări
- We498 Flyer FinalDocument1 paginăWe498 Flyer Finalsami_bangash_1Încă nu există evaluări
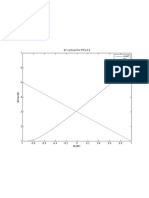
- 7 B1 Vs Exact For P/Pe:0.8 Exact B1Document1 pagină7 B1 Vs Exact For P/Pe:0.8 Exact B1sami_bangash_1Încă nu există evaluări
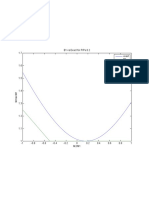
- 1.7 B1 Vs Exact For P/Pe:0.2 Exact B1Document1 pagină1.7 B1 Vs Exact For P/Pe:0.2 Exact B1sami_bangash_1Încă nu există evaluări
- Masonry Photographic Study TourDocument1 paginăMasonry Photographic Study Toursami_bangash_1Încă nu există evaluări
- Nonlinear Steel Assignment TwoDocument2 paginiNonlinear Steel Assignment Twosami_bangash_1Încă nu există evaluări
- Comparison: Euler 2 Order Elastic 2 Order Elastic NRM (Mod) 2 Order Elastic NRM (Ideal) 2 Order Inelastic Linear ElasticDocument1 paginăComparison: Euler 2 Order Elastic 2 Order Elastic NRM (Mod) 2 Order Elastic NRM (Ideal) 2 Order Inelastic Linear Elasticsami_bangash_1Încă nu există evaluări
- Rebar Pullout Testing PDFDocument6 paginiRebar Pullout Testing PDFsami_bangash_1Încă nu există evaluări
- Manual de Instruções John Deere D170 (56 Páginas)Document2 paginiManual de Instruções John Deere D170 (56 Páginas)Antonio CostaÎncă nu există evaluări
- Kathrein 739506Document2 paginiKathrein 739506Carlos CostaÎncă nu există evaluări
- Grid Synchronization of Power Converters Using Multiple Second Order Generalized IntegratorsDocument6 paginiGrid Synchronization of Power Converters Using Multiple Second Order Generalized IntegratorsJandfor Tansfg ErrottÎncă nu există evaluări
- Presented By:: Rafi Sheikh Sheeraz Malik Syed Ahmed Ali Umair Ali Waqar AmeenDocument34 paginiPresented By:: Rafi Sheikh Sheeraz Malik Syed Ahmed Ali Umair Ali Waqar AmeenSyed Ahmed AliÎncă nu există evaluări
- Functions of An EngineerDocument5 paginiFunctions of An EngineerDEUS PHILIP DURANÎncă nu există evaluări
- Astm A769 PDFDocument5 paginiAstm A769 PDFCristian OtivoÎncă nu există evaluări
- Home Automation Control System Using DTMFDocument25 paginiHome Automation Control System Using DTMFengaydiÎncă nu există evaluări
- Chemical Induetries-1Document75 paginiChemical Induetries-1Muhammad Anees Ur RehmanÎncă nu există evaluări
- Antena 700 2m - TongyuDocument2 paginiAntena 700 2m - TongyuLenin Alejandro Ramirez HuaypatinÎncă nu există evaluări
- DUNAN Fan Coil UnitDocument14 paginiDUNAN Fan Coil UnitDjordjeÎncă nu există evaluări
- X2 / 275 Vac: B 81 191 EMI Suppression CapacitorsDocument4 paginiX2 / 275 Vac: B 81 191 EMI Suppression CapacitorsMeg YorkÎncă nu există evaluări
- SAX Brochure - Web ReadyDocument4 paginiSAX Brochure - Web ReadyEng-Ahmad Abo-AledousÎncă nu există evaluări
- MTS Mto Ato Cto Eto PDFDocument5 paginiMTS Mto Ato Cto Eto PDFJuan Villanueva ZamoraÎncă nu există evaluări
- DNF Stand PipeDocument3 paginiDNF Stand PipeChristopher BrownÎncă nu există evaluări
- Tools of BiotechnologyDocument10 paginiTools of Biotechnologyiamforu1Încă nu există evaluări
- Exception Handling Notes For Vtu StudentsDocument42 paginiException Handling Notes For Vtu StudentshelloÎncă nu există evaluări
- db2v9 EsqlDocument419 paginidb2v9 EsqlRaphael PugliesiÎncă nu există evaluări
- Lab Gas FlowmeterDocument7 paginiLab Gas Flowmeterazym94Încă nu există evaluări
- Computer Organization: - by Rama Krishna Thelagathoti (M.Tech CSE From IIT Madras)Document118 paginiComputer Organization: - by Rama Krishna Thelagathoti (M.Tech CSE From IIT Madras)iamy2ramsÎncă nu există evaluări
- Stelzer Catalog PDFDocument12 paginiStelzer Catalog PDFlhphong021191Încă nu există evaluări
- Cover Letter Mechanical Engineering Nondestructive TestingDocument1 paginăCover Letter Mechanical Engineering Nondestructive TestingGagandeep SinghÎncă nu există evaluări
- Key Features Boilermaker Proven, Legendary ReliabilityDocument2 paginiKey Features Boilermaker Proven, Legendary ReliabilityManuel Cantoral CortazarÎncă nu există evaluări
- Manual Deus II UkDocument52 paginiManual Deus II UkMariusÎncă nu există evaluări
- 2015 VGP Checklist - Rev0Document9 pagini2015 VGP Checklist - Rev0Takis RappasÎncă nu există evaluări
- Toshiba 42pw33q - S Ch. Pw33 SchematicsDocument18 paginiToshiba 42pw33q - S Ch. Pw33 SchematicstodorloncarskiÎncă nu există evaluări