S-ar putea să vă placă și
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (587)
- The Comparison of Methods To Assess in Vitro Drug Release From NanoparticlesDocument1 paginăThe Comparison of Methods To Assess in Vitro Drug Release From NanoparticlesLuciaGomezÎncă nu există evaluări
- Contact Dermatitis 2015parameterDocument39 paginiContact Dermatitis 2015parameterSaniya Ilma ArifaÎncă nu există evaluări
- Haemophilus Influenzae Disease ManualDocument12 paginiHaemophilus Influenzae Disease ManualLuciaGomezÎncă nu există evaluări
- Drug Release Testing Methods of Polymeric Particulate Drug FormulationsDocument9 paginiDrug Release Testing Methods of Polymeric Particulate Drug FormulationsLuciaGomezÎncă nu există evaluări
- Stool AnalysisDocument8 paginiStool AnalysisLuciaGomez100% (1)
- Farmacología CrashDocument236 paginiFarmacología CrashGonzalo Sterling100% (1)
- Metabolic Syndrome Diabetes and Hyperuricemia PDFDocument7 paginiMetabolic Syndrome Diabetes and Hyperuricemia PDFLuciaGomezÎncă nu există evaluări
- Topical Menthol On Vascular Responses and Muscular ContractionsDocument6 paginiTopical Menthol On Vascular Responses and Muscular ContractionsLuciaGomezÎncă nu există evaluări
- The Metabolic Syndrome and HivDocument8 paginiThe Metabolic Syndrome and HivLuciaGomezÎncă nu există evaluări
- Diabetes and AlzheimerDocument8 paginiDiabetes and AlzheimerLuciaGomezÎncă nu există evaluări
- Potential Health Benefits of Conjugated Linoleic Acid (CLA)Document14 paginiPotential Health Benefits of Conjugated Linoleic Acid (CLA)LuciaGomezÎncă nu există evaluări
- How Can Fungi Improve Paper Manufacturing?Document2 paginiHow Can Fungi Improve Paper Manufacturing?LuciaGomezÎncă nu există evaluări
- Diabetes During PregnancyDocument44 paginiDiabetes During PregnancyLuciaGomezÎncă nu există evaluări
- Risk Test Paper Version PDFDocument2 paginiRisk Test Paper Version PDFLuciaGomezÎncă nu există evaluări
- AmebaeDocument10 paginiAmebaeLuciaGomezÎncă nu există evaluări
- Chapter 20Document41 paginiChapter 20Amer RahmahÎncă nu există evaluări
- Meal and Animal NutritionDocument1 paginăMeal and Animal NutritionLuciaGomezÎncă nu există evaluări
- Hulley. Designing Clinical Research. 2nd EdDocument328 paginiHulley. Designing Clinical Research. 2nd EdAlfonso Fernandez Pazos100% (1)
- EPA and DHA ExtractionDocument12 paginiEPA and DHA ExtractionLuciaGomezÎncă nu există evaluări
- Antimicrobial Peptides of Probiotic Lactobacillus StrainsDocument5 paginiAntimicrobial Peptides of Probiotic Lactobacillus StrainsLuciaGomezÎncă nu există evaluări
- Biodeterioration of Paper (Fungi)Document10 paginiBiodeterioration of Paper (Fungi)LuciaGomez0% (1)
- Centrifugation HandoutDocument3 paginiCentrifugation HandoutLuciaGomezÎncă nu există evaluări
- Arteries & Veins of The Upper LimbDocument4 paginiArteries & Veins of The Upper LimbPirabakar MahendranÎncă nu există evaluări
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (119)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- Pharmaceutics 09 00041 v2Document14 paginiPharmaceutics 09 00041 v2thasyaÎncă nu există evaluări
- Q and A DactylosDocument56 paginiQ and A DactylosJUNN REE MONTILLA100% (2)
- Topic Test Oxfordaqa Int Gcse Biology 9201 BioenergeticsDocument29 paginiTopic Test Oxfordaqa Int Gcse Biology 9201 BioenergeticsNovanolo Christovori ZebuaÎncă nu există evaluări
- Excerpt From "Presence" by Amy CuddyDocument10 paginiExcerpt From "Presence" by Amy CuddyOnPointRadioÎncă nu există evaluări
- Life ProcessesDocument53 paginiLife Processessagar100% (1)
- Evolutionary Reclassification of Protozoa and Chromista KingdomsDocument4 paginiEvolutionary Reclassification of Protozoa and Chromista KingdomsWormInchÎncă nu există evaluări
- Postpartum HemorrhageDocument25 paginiPostpartum HemorrhageaKmaL67% (3)
- Drug 1Document2 paginiDrug 1Nicholas TagleÎncă nu există evaluări
- Alaryngeal SpeechDocument1 paginăAlaryngeal Speechvee propagandaÎncă nu există evaluări
- Platelet Rich Plasma in OrthopaedicsDocument269 paginiPlatelet Rich Plasma in OrthopaedicsBelinda Azhari SiswantoÎncă nu există evaluări
- Head Toe Physical AssessmentDocument2 paginiHead Toe Physical Assessmentzbestgurl100% (2)
- Rhopalocera (Butterfly) : FunctionsDocument18 paginiRhopalocera (Butterfly) : FunctionsChris Anthony EdulanÎncă nu există evaluări
- Use of Vasopressors and Inotropes - UpToDateDocument25 paginiUse of Vasopressors and Inotropes - UpToDateVictor Mendoza - MendezÎncă nu există evaluări
- Pathologyofdiseasesof Geriatricexoticmammals: Drury R. Reavill,, Denise M. ImaiDocument34 paginiPathologyofdiseasesof Geriatricexoticmammals: Drury R. Reavill,, Denise M. ImaiRaquel MotaÎncă nu există evaluări
- ECG Interpretation BookDocument57 paginiECG Interpretation BookLouis Plan100% (2)
- Review of System Done... 1Document2 paginiReview of System Done... 1Dheng EsquijoÎncă nu există evaluări
- MSDS Garam MejaDocument5 paginiMSDS Garam MejaDesyrulaÎncă nu există evaluări
- ACE Biology O'level Book@2021Document213 paginiACE Biology O'level Book@2021DuyÎncă nu există evaluări
- OADocument27 paginiOADarkKnighthere100% (1)
- Ncps For CvaDocument14 paginiNcps For Cvalouie roderos0% (2)
- NKC Fast Facts - Poop Chart - 5 2017 PDFDocument1 paginăNKC Fast Facts - Poop Chart - 5 2017 PDFFaniaÎncă nu există evaluări
- Microsoft Word - Endodontic - MishapsDocument20 paginiMicrosoft Word - Endodontic - MishapsShufeiÎncă nu există evaluări
- In This Issue: Wound Infection and ColonisationDocument7 paginiIn This Issue: Wound Infection and ColonisationSeftiana WahyuniÎncă nu există evaluări
- Mphil Bio-Chemistry ProjectDocument98 paginiMphil Bio-Chemistry ProjectBalaji Rao NÎncă nu există evaluări
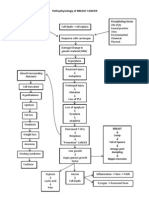
- Pathophysiology of BREAST CANCERDocument1 paginăPathophysiology of BREAST CANCERAlinor Abubacar100% (6)
- Application of Proper Draping-Final DemoDocument15 paginiApplication of Proper Draping-Final DemoMarife Hernandez Gelin80% (15)
- Reflective Writing in MedicineDocument10 paginiReflective Writing in MedicineMÎncă nu există evaluări
- Advanced Trauma Life SupportDocument452 paginiAdvanced Trauma Life Supportnatalia100% (7)
- Neuroscience Pathways Fall 2012Document46 paginiNeuroscience Pathways Fall 2012Yezin ShamoonÎncă nu există evaluări
- Asthma Care Quick Reference - Diagnosing and Managing AsthmaDocument18 paginiAsthma Care Quick Reference - Diagnosing and Managing AsthmaMarizka Putri AftriaÎncă nu există evaluări