S-ar putea să vă placă și
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleDe la EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleÎncă nu există evaluări
- Hipertensión Pulmonar Arterial: (Ascitis) En Pollos De EngordaDe la EverandHipertensión Pulmonar Arterial: (Ascitis) En Pollos De EngordaÎncă nu există evaluări
- Villamemo - RM 2022 - HematologíaDocument20 paginiVillamemo - RM 2022 - HematologíaGab VergaraÎncă nu există evaluări
- Hematologia Preguntas 1 2004-2005Document37 paginiHematologia Preguntas 1 2004-2005gerispice18100% (1)
- HEMATOLOGIA - Hemograma Completo - Checked A ImprimerDocument25 paginiHEMATOLOGIA - Hemograma Completo - Checked A ImprimerGUillaume100% (1)
- Revision Mielonisis Central PontinaDocument4 paginiRevision Mielonisis Central PontinaDavid ColqueÎncă nu există evaluări
- Guia de Estudio HematologiaDocument11 paginiGuia de Estudio HematologiaSandraGalindoZevallosÎncă nu există evaluări
- Hematología Pediátrica CRUDocument46 paginiHematología Pediátrica CRUmohamed salah100% (1)
- Actualizacion en El Diagnóstico y Manejo de Las HepatopatiasDocument6 paginiActualizacion en El Diagnóstico y Manejo de Las HepatopatiasSara LeeÎncă nu există evaluări
- Algoritmos en Nefrologia Modulo 2Document34 paginiAlgoritmos en Nefrologia Modulo 2norma paulina carcausto lipaÎncă nu există evaluări
- 03.009 Protocolo Diagnóstico de Las Anemias HemolíticasDocument2 pagini03.009 Protocolo Diagnóstico de Las Anemias HemolíticasGustavo AraujoÎncă nu există evaluări
- Casos ClinicosDocument36 paginiCasos ClinicosAlejandra MonteroÎncă nu există evaluări
- Fibrilación Auricular BraunwaldDocument23 paginiFibrilación Auricular BraunwaldJuan GarciaÎncă nu există evaluări
- Perlas Clinicas Anemia HemoliticaDocument2 paginiPerlas Clinicas Anemia HemoliticaDra Mary PalominoÎncă nu există evaluări
- Ideas Clave HemetologiaDocument26 paginiIdeas Clave HemetologiaFroylan GalindoÎncă nu există evaluări
- NEUROLOGÍADocument88 paginiNEUROLOGÍAgrafeÎncă nu există evaluări
- Hemorragia Digestiva SuperiorDocument5 paginiHemorragia Digestiva SuperiorAriana Calzadilla RuizÎncă nu există evaluări
- IRCDocument10 paginiIRCGerardo Pabón GallinoÎncă nu există evaluări
- Resumen MI Nefro Completo PDFDocument25 paginiResumen MI Nefro Completo PDFLuis F Concha ManriquezÎncă nu există evaluări
- Minilibro MedintDocument108 paginiMinilibro MedintanoÎncă nu există evaluări
- GuacalaDocument2 paginiGuacalaArsenio Flores VasquezÎncă nu există evaluări
- Parcial Medicina InternaDocument14 paginiParcial Medicina InternaJosè BermúdezÎncă nu există evaluări
- La Importancia Del Frotis de Sangre PeriféricaDocument16 paginiLa Importancia Del Frotis de Sangre PeriféricaSilvano keny Falcon BerrospiÎncă nu există evaluări
- Fármacos Antiarrítmicos: de la Teoría a la PrácticaDocument31 paginiFármacos Antiarrítmicos: de la Teoría a la PrácticaCamila AguilaÎncă nu există evaluări
- Manifestaciones Cutáneas de Linfomas y LeucimiasDocument66 paginiManifestaciones Cutáneas de Linfomas y LeucimiasinnovaciondocenteÎncă nu există evaluări
- Unas Preguntas para Estudiar para El Segundo Examen Parcial de HematologíaDocument7 paginiUnas Preguntas para Estudiar para El Segundo Examen Parcial de HematologíaSamiri SamiriÎncă nu există evaluări
- Cirrosis HepaticaDocument52 paginiCirrosis HepaticaDokhur100% (1)
- Endocrinologia Proedumed 3Document11 paginiEndocrinologia Proedumed 3Kariina LarreguiiÎncă nu există evaluări
- Anemia MegaloblásticaDocument31 paginiAnemia MegaloblásticaLizbeth RomeroÎncă nu există evaluări
- Anemia Por Deficiencia de Acido Folico Parte2Document10 paginiAnemia Por Deficiencia de Acido Folico Parte2Lis LisÎncă nu există evaluări
- Test Alto Rendimiento Endocrinologia PDFDocument24 paginiTest Alto Rendimiento Endocrinologia PDFChris GreenÎncă nu există evaluări
- Toxinas Uremicas TrabajoDocument2 paginiToxinas Uremicas TrabajoAlbertoServin100% (1)
- Anemia AplásicaDocument5 paginiAnemia AplásicaNEYDA QUISPE CARHUANIÎncă nu există evaluări
- Exarmed FlashcardsDocument30 paginiExarmed FlashcardsRodrigo FonsecaÎncă nu există evaluări
- HiponatremiaDocument8 paginiHiponatremiaMiguel Espinosa SaenzÎncă nu există evaluări
- Dislipemias AmfDocument12 paginiDislipemias AmfMiguel RobresÎncă nu există evaluări
- Etiología, Clasificación y Epidemiología Del Accidente Cerebrovascular - UpToDateDocument18 paginiEtiología, Clasificación y Epidemiología Del Accidente Cerebrovascular - UpToDateAnita Rocio Salas CastroÎncă nu există evaluări
- Eunacom Hematología 2014 PDFDocument136 paginiEunacom Hematología 2014 PDFAmalia MoralesÎncă nu există evaluări
- Manejo de Hiponatremia 2022Document20 paginiManejo de Hiponatremia 2022Guissela Montoya LopezÎncă nu există evaluări
- Preguntas HematologíaDocument6 paginiPreguntas Hematologíarrrm1968100% (1)
- Oftalmología MIR 2004/2005: Enfermedades orbitarias y externasDocument17 paginiOftalmología MIR 2004/2005: Enfermedades orbitarias y externasANGUIE ANETTE MAMANI SUCASAIREÎncă nu există evaluări
- HipertrigliceridemiaDocument15 paginiHipertrigliceridemiaROSE SASCKA CHAVEZÎncă nu există evaluări
- Mieloma Multiple TratamientoDocument80 paginiMieloma Multiple TratamientoAlexis VegaÎncă nu există evaluări
- 06 SX MieloproliferativosDocument15 pagini06 SX MieloproliferativosSandy Salazar100% (1)
- Insuficiencia Renal AgudaDocument41 paginiInsuficiencia Renal AgudaEstefanía P. HerreraÎncă nu există evaluări
- Resumen NefrologíaDocument36 paginiResumen NefrologíaYasna Cabello MuñozÎncă nu există evaluări
- Betabloqueadores en HTA 2018Document69 paginiBetabloqueadores en HTA 2018NICOLASÎncă nu există evaluări
- ParacentesisDocument4 paginiParacentesisJosué Ajsr100% (1)
- Anemia HemoliticaDocument78 paginiAnemia Hemoliticanatalia_AS166615Încă nu există evaluări
- Mieloma y RiñónDocument44 paginiMieloma y RiñónnefrovillanuevaÎncă nu există evaluări
- Interpretacion Gasometrica RazaDocument8 paginiInterpretacion Gasometrica RazaEnrique Vazquez RodriguezÎncă nu există evaluări
- Cuestionario Sindromes Mieloproliferativos 13junio 2014 RosaDocument3 paginiCuestionario Sindromes Mieloproliferativos 13junio 2014 RosaRosa AbastaÎncă nu există evaluări
- Fisiopatologia de Glomerulopatias 2014Document51 paginiFisiopatologia de Glomerulopatias 2014LilianaCondor100% (1)
- Reumatología Clinica PDFDocument3 paginiReumatología Clinica PDFgvargasmÎncă nu există evaluări
- Trombocitopenia y PancitopeniaDocument31 paginiTrombocitopenia y PancitopeniaHugo Maximiliano Solis Aguayo100% (1)
- MECANISMOS INMUNOLÓGICOS Y CELULARES DE LA LESIÓN GLOMERULARDocument13 paginiMECANISMOS INMUNOLÓGICOS Y CELULARES DE LA LESIÓN GLOMERULARChristian Itandehui JiménezÎncă nu există evaluări
- Síndrome Mononucleósico: Características Clínicas y de LaboratorioDocument91 paginiSíndrome Mononucleósico: Características Clínicas y de LaboratorioRodrigoÎncă nu există evaluări
- Anemia AplásicaDocument33 paginiAnemia AplásicaJason Trevejo CruzÎncă nu există evaluări
- Integración funcional del celador en el área de urgenciasDe la EverandIntegración funcional del celador en el área de urgenciasÎncă nu există evaluări
- KawasakiDocument7 paginiKawasakiAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- 13 Enfermedad KawasakiDocument13 pagini13 Enfermedad KawasakiFernanda MartinezÎncă nu există evaluări
- Tabla Farmacos 2Document9 paginiTabla Farmacos 2Andrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Eunacom 2010Document59 paginiEunacom 2010urbanincultureÎncă nu există evaluări
- Algoritmos de Cifuentes en Ginecología y Obstetricia PDFDocument406 paginiAlgoritmos de Cifuentes en Ginecología y Obstetricia PDFYhulissa Chavez100% (1)
- Algoritmos Pediatria Gabriela AmayaDocument4 paginiAlgoritmos Pediatria Gabriela AmayaAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Neurologia PDFDocument135 paginiNeurologia PDFAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Programa de Salud de La Mujer 1997Document170 paginiPrograma de Salud de La Mujer 1997gersonito7Încă nu există evaluări
- DOCUMENTOCOMISIONDocument26 paginiDOCUMENTOCOMISIONAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Guia AEMIR de Actuacion en Urgencias PDFDocument310 paginiGuia AEMIR de Actuacion en Urgencias PDFAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Algoritmos en Nefrologia SENDocument380 paginiAlgoritmos en Nefrologia SENviaereaÎncă nu există evaluări
- Manual de Medicina InternaDocument145 paginiManual de Medicina InternaAaron Elvir100% (1)
- Resumen Ada 2014Document22 paginiResumen Ada 2014Andrea Carolina Carrasco Rodriguez100% (1)
- Notas de Pediatria PDFDocument265 paginiNotas de Pediatria PDFleidyÎncă nu există evaluări
- Mallas Reparacion Pared AbdominalDocument6 paginiMallas Reparacion Pared AbdominalAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- DermatopatologíaDocument61 paginiDermatopatologíaAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
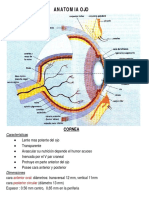
- Anatomía OcularDocument51 paginiAnatomía OcularAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Atencion PrimariaDocument142 paginiAtencion PrimariaAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Cuadros de MeningitisDocument1 paginăCuadros de MeningitisAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Dubin Interpretacion de ECGIMPRIMIRDocument64 paginiDubin Interpretacion de ECGIMPRIMIRAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Guias Alad 2013 1Document142 paginiGuias Alad 2013 1wipi112Încă nu există evaluări
- EcgDocument12 paginiEcgAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Resumen Dm2 AladDocument24 paginiResumen Dm2 AladAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Ingredientes Kuchen de ManzanaDocument2 paginiIngredientes Kuchen de ManzanaAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Tto y Rofilaxis Neumonia VihDocument3 paginiTto y Rofilaxis Neumonia VihAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Respiratorio 2014Document162 paginiRespiratorio 2014Andrea Carolina Carrasco RodriguezÎncă nu există evaluări
- EPOCDocument5 paginiEPOCAndrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Tumores y Lesiones Pseudoumorales de Partes Blandas-2Document58 paginiTumores y Lesiones Pseudoumorales de Partes Blandas-2Andrea Carolina Carrasco RodriguezÎncă nu există evaluări
- Osteologia de ExtremidadesDocument13 paginiOsteologia de ExtremidadesMariana Arroyo RamírezÎncă nu există evaluări
- Dermatomiositis 2Document37 paginiDermatomiositis 2Melissa CantilloÎncă nu există evaluări
- Panorama Epidemiológico de La Infancia y A La Adolescencia en MéxicoDocument40 paginiPanorama Epidemiológico de La Infancia y A La Adolescencia en MéxicoCarlos Baas AkeÎncă nu există evaluări
- Malformaciones de La PróstataDocument12 paginiMalformaciones de La PróstataSimone SeguraÎncă nu există evaluări
- Pólipos Del Colon y RectoDocument8 paginiPólipos Del Colon y RectoDiego Mogrovejo MolinaÎncă nu există evaluări
- Exposición - Cancer GástricoDocument5 paginiExposición - Cancer GástricoKassandra C LucanaÎncă nu există evaluări
- Linfoma de HodgkinDocument57 paginiLinfoma de HodgkinKenny Alberto Melendres QuispeÎncă nu există evaluări
- Distocia 160716014242Document10 paginiDistocia 160716014242Livia PinedaÎncă nu există evaluări
- Barreras Celulares Del Sistema InnatoDocument58 paginiBarreras Celulares Del Sistema InnatoEvelyn Neira100% (2)
- Control 7 QuimicaDocument9 paginiControl 7 QuimicaKarem Alejandra Rojas AlfaroÎncă nu există evaluări
- DentavoxDocument303 paginiDentavoxGenesiisMariñoÎncă nu există evaluări
- URINOTERAPIADocument17 paginiURINOTERAPIAJHERAL ALEXIS SURICHAQUI GARCIAÎncă nu există evaluări
- Beneficios Del ResveratrolDocument7 paginiBeneficios Del ResveratrolMilkyReitxuÎncă nu există evaluări
- Regulación de la expresión génica mediada por nutrientes y componentes alimentariosDocument42 paginiRegulación de la expresión génica mediada por nutrientes y componentes alimentariosLupita100% (1)
- Ictericia Obstructiva Benigna FinalDocument38 paginiIctericia Obstructiva Benigna Finaljorge andres100% (1)
- Ecografia de Bazo y PancreasDocument21 paginiEcografia de Bazo y PancreaskendyblueÎncă nu există evaluări
- Cuidados Pos Operatorio y TrasladoDocument21 paginiCuidados Pos Operatorio y TrasladoJoaquin GuzmánÎncă nu există evaluări
- Historia Clinica FacialDocument4 paginiHistoria Clinica FacialkarenÎncă nu există evaluări
- Laboratorio Clínico - Marcadores TumoralesDocument19 paginiLaboratorio Clínico - Marcadores TumoralesUSMP FN ARCHIVOS100% (1)
- El PNI (Positivo, Negarivo, Interesante)Document4 paginiEl PNI (Positivo, Negarivo, Interesante)LuisAldanadeLeónÎncă nu există evaluări
- Funciones de La Interleucina 8Document5 paginiFunciones de La Interleucina 8Ron JimÎncă nu există evaluări
- Amputacion UAEMDocument113 paginiAmputacion UAEMEliasBandaNavaÎncă nu există evaluări
- Salud Ocupacional y Enfermedades Relacionadas Con El TrabajoDocument41 paginiSalud Ocupacional y Enfermedades Relacionadas Con El TrabajoOscar David Bellido ZarateÎncă nu există evaluări
- Regimen OshawaDocument2 paginiRegimen Oshawaronanpg100% (2)
- Presentacion Hepatitis B, AmparoDocument31 paginiPresentacion Hepatitis B, AmparoRoly2013Încă nu există evaluări
- Presentación NonicotDocument16 paginiPresentación Nonicotangelmoncayo7695100% (1)
- Toxicidad en Materiales.Document25 paginiToxicidad en Materiales.Franco Mauricio100% (2)
- Historia ClinicaDocument32 paginiHistoria ClinicaAngelita Suki KhanÎncă nu există evaluări
- Tumores 1Document22 paginiTumores 1Fransheska Mendoza LunaÎncă nu există evaluări
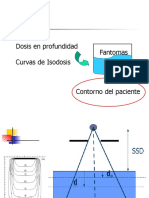
- Planificación Tratamientos RADIOTERAPIADocument75 paginiPlanificación Tratamientos RADIOTERAPIASaskyaCatalinaCuelloLobosÎncă nu există evaluări