S-ar putea să vă placă și
- Asean Guidelines For: Final Draft: 21 July 2004Document29 paginiAsean Guidelines For: Final Draft: 21 July 2004Jone YingÎncă nu există evaluări
- ASEAN BE Guidelines-Amended According To1st BABE TWG15th PPWGDocument29 paginiASEAN BE Guidelines-Amended According To1st BABE TWG15th PPWGTarikÎncă nu există evaluări
- Note For Guidance On The Investigation of Bioavailability & BioequivalenceDocument19 paginiNote For Guidance On The Investigation of Bioavailability & BioequivalenceAhmed AliÎncă nu există evaluări
- Guideline On Bioavailability and BioequivalanceDocument26 paginiGuideline On Bioavailability and BioequivalanceGima Amezia SariÎncă nu există evaluări
- Bioequevalance StudiesDocument55 paginiBioequevalance StudiesMubammad MursaleenÎncă nu există evaluări
- Bioavailability and BioequivalenceDocument28 paginiBioavailability and Bioequivalencekulbhushan singhÎncă nu există evaluări
- Bioavailability and BioequivalenceDocument6 paginiBioavailability and BioequivalenceDharmesh PatelÎncă nu există evaluări
- Hemanagadurga2019 Bioavailability StudiesDocument17 paginiHemanagadurga2019 Bioavailability Studiesraghuraj75Încă nu există evaluări
- Drug Product Performance GuideDocument119 paginiDrug Product Performance GuideSachin BagewadiÎncă nu există evaluări
- Nomenclature terms for therapeutic equivalenceDocument4 paginiNomenclature terms for therapeutic equivalencekurutalaÎncă nu există evaluări
- Final ProjectDocument119 paginiFinal ProjectSaurabh KumarÎncă nu există evaluări
- Bioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyDocument15 paginiBioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyMayson Bali100% (1)
- Review Article: Barbara Davit, April C. Braddy, Dale P. Conner, and Lawrence X. YuDocument17 paginiReview Article: Barbara Davit, April C. Braddy, Dale P. Conner, and Lawrence X. Yulhthang1990Încă nu există evaluări
- Bioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyDocument16 paginiBioequivalence: Faculty of Pharmacy, Nursing and Health Professions Master in Industrial Pharmaceutical TechnologyMayson BaliÎncă nu există evaluări
- Ba-Be PDFDocument30 paginiBa-Be PDFUswatun Hasanah7201Încă nu există evaluări
- Guideline For Bioequivalence Studies of Generic ProductsDocument23 paginiGuideline For Bioequivalence Studies of Generic ProductschetanjmistryÎncă nu există evaluări
- 8 Ba Be FdaDocument36 pagini8 Ba Be Fdaraju narayana padalaÎncă nu există evaluări
- IntroductionDocument71 paginiIntroductionAreenub ArshadÎncă nu există evaluări
- KjbvggvvvDocument10 paginiKjbvggvvvSuvojit BasakÎncă nu există evaluări
- Bioequivalencestudies 2Document16 paginiBioequivalencestudies 2Aamir NawazÎncă nu există evaluări
- BIOEQUI2Document5 paginiBIOEQUI2Laiba KhalidÎncă nu există evaluări
- Drug Delivery Inhalation BioequivalenceDocument12 paginiDrug Delivery Inhalation Bioequivalenceparinaferns0% (1)
- Lecture 10 - PHR514-Pharmacy Law and Regulatory AffairsDocument26 paginiLecture 10 - PHR514-Pharmacy Law and Regulatory AffairsNeymar ShuvoÎncă nu există evaluări
- Requirement of Dissolution Test For f2 - Gastro Resistant TabletDocument17 paginiRequirement of Dissolution Test For f2 - Gastro Resistant Tabletfad12345Încă nu există evaluări
- Regulatory Perspective on Bioequivalence StudiesDocument12 paginiRegulatory Perspective on Bioequivalence Studiesbhanu99100% (2)
- 24 13v3i2 5Document9 pagini24 13v3i2 5gopalÎncă nu există evaluări
- Definition of BiopharmaceuticsDocument3 paginiDefinition of BiopharmaceuticsvafaashkÎncă nu există evaluări
- Bio Availability and Bioequivalane-LastDocument13 paginiBio Availability and Bioequivalane-LastAhmedothman62100% (1)
- Article On BA&BEDocument7 paginiArticle On BA&BENitin DhimanÎncă nu există evaluări
- Bioavailability and BioequivalenceDocument56 paginiBioavailability and Bioequivalenceنور الهدى100% (1)
- Assignment - BioequivalenceDocument24 paginiAssignment - BioequivalenceSM Sabuj AfridiÎncă nu există evaluări
- Bioavailability and Bieq Review ArticleDocument15 paginiBioavailability and Bieq Review ArticlevinayÎncă nu există evaluări
- Clinical Pharmacokinetic Studies of PharmaceuticalsDocument23 paginiClinical Pharmacokinetic Studies of PharmaceuticalsPankaj SharmaÎncă nu există evaluări
- Difference Between Bioavailability and BioequivalenceDocument11 paginiDifference Between Bioavailability and Bioequivalenceprakash deshmukhÎncă nu există evaluări
- Registration of Medicines Dissolution GuidelineDocument11 paginiRegistration of Medicines Dissolution GuidelineHarrcanaa RajahÎncă nu există evaluări
- What Is Biopharmaceutics?: Brand NameDocument10 paginiWhat Is Biopharmaceutics?: Brand NameAhmad Jamal HashmiÎncă nu există evaluări
- Bioavailability and Bioequivalence (BABE)Document4 paginiBioavailability and Bioequivalence (BABE)Wingielyn Honculada BaldozaÎncă nu există evaluări
- Regulatory Requirement of Similar Biologic For Marketing AuthorizationDocument19 paginiRegulatory Requirement of Similar Biologic For Marketing AuthorizationSatheesh SachinÎncă nu există evaluări
- Bioavailability and BioequivalenceDocument47 paginiBioavailability and BioequivalenceGiovanne BuendiaÎncă nu există evaluări
- Bioequivelance StudiesDocument16 paginiBioequivelance StudiesJayÎncă nu există evaluări
- Bioanalysis NotesDocument7 paginiBioanalysis NoteskeerthiÎncă nu există evaluări
- AmlodipinebesylatepaperDocument6 paginiAmlodipinebesylatepaperRakesh RauniyarÎncă nu există evaluări
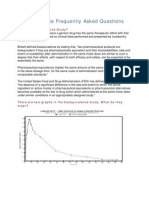
- Bioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Document4 paginiBioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Ahmed HasanÎncă nu există evaluări
- Drug Absorption PDFDocument117 paginiDrug Absorption PDFAndhia DhiyaÎncă nu există evaluări
- Pkin08 Soal Biofar Pak BoesroDocument117 paginiPkin08 Soal Biofar Pak Boesroabu_59Încă nu există evaluări
- Pharmaceutics 14 02565Document12 paginiPharmaceutics 14 02565Sebas IsraelÎncă nu există evaluări
- Bio Similar S GuidanceDocument6 paginiBio Similar S Guidancegulafsha1Încă nu există evaluări
- Register biosimilar biological products SingaporeDocument13 paginiRegister biosimilar biological products SingaporeWilliam ChandraÎncă nu există evaluări
- The Orange BookDocument6 paginiThe Orange Book50KMKDIVYA RAJPALÎncă nu există evaluări
- THEORANGEBOOKDocument6 paginiTHEORANGEBOOKMashiko DakhundaridzeÎncă nu există evaluări
- Bioavailability and BioequivalenceDocument39 paginiBioavailability and Bioequivalenceنور الهدىÎncă nu există evaluări
- Guided Equivalence StudyDocument30 paginiGuided Equivalence StudyTejas PatelÎncă nu există evaluări
- Pre 66222Document18 paginiPre 66222Md. JewelÎncă nu există evaluări
- Psychotropic DrugDocument10 paginiPsychotropic DrugAlexandra Elizabeth FloresÎncă nu există evaluări
- Usp 1151Document10 paginiUsp 1151Karnati PraveenaÎncă nu există evaluări
- Bioavailability and BioequivalenceDocument81 paginiBioavailability and BioequivalenceNiharika ModiÎncă nu există evaluări
- Biosimilars in Ophthalmology: A ReviewDocument11 paginiBiosimilars in Ophthalmology: A Reviewindex PubÎncă nu există evaluări
- Update On Gastrointestinal Biorelevant Media and Physiologically Relevant Dissolution ConditionsDocument14 paginiUpdate On Gastrointestinal Biorelevant Media and Physiologically Relevant Dissolution ConditionsVirajÎncă nu există evaluări
- Biological Aspects of FormulationsDocument8 paginiBiological Aspects of FormulationsKabirÎncă nu există evaluări
- TOEFL NotesDocument2 paginiTOEFL NotesDianeÎncă nu există evaluări
- UTIDocument46 paginiUTIGlorya Benthamy SiamiloyÎncă nu există evaluări
- Indonesia: Applying For An Australia Awards ScholarshipDocument4 paginiIndonesia: Applying For An Australia Awards ScholarshipSarah MuharomahÎncă nu există evaluări
- Barthel Index (0-20)Document2 paginiBarthel Index (0-20)Yessika Adelwin Natalia95% (19)
- UTIDocument27 paginiUTIDianeÎncă nu există evaluări
- GGGGG GGGGG GGGGG GGGGG GGGGGDocument1 paginăGGGGG GGGGG GGGGG GGGGG GGGGGDianeÎncă nu există evaluări
- Urinary Tract Infection (UTI)Document38 paginiUrinary Tract Infection (UTI)Trixie Anne GamotinÎncă nu există evaluări
- The 2015 Bls Acls Guidelines EnaDocument36 paginiThe 2015 Bls Acls Guidelines EnaAstriTaufiRamadhaniÎncă nu există evaluări
- Enzyme Crossword: Key Terms for Enzyme ActivityDocument3 paginiEnzyme Crossword: Key Terms for Enzyme ActivityormattÎncă nu există evaluări
- Pharmacology NotesDocument2 paginiPharmacology Notessakuragi jakeÎncă nu există evaluări
- Marcel Dekker SeriesDocument11 paginiMarcel Dekker Seriesmaulikpatel27090% (1)
- Dr. Widyati ADR ANALYSIS-PERSIDocument39 paginiDr. Widyati ADR ANALYSIS-PERSIHanaÎncă nu există evaluări
- DTH Pharmacy PresentationDocument46 paginiDTH Pharmacy PresentationdrkefyalewtayeÎncă nu există evaluări
- How Knowledge About ADRs Is CreatedDocument17 paginiHow Knowledge About ADRs Is CreatedAnonymous whcvnPBeQÎncă nu există evaluări
- Egyptian Guidelines For Good Clinical Practice (GCP) On Pharmaceutical ProductsDocument59 paginiEgyptian Guidelines For Good Clinical Practice (GCP) On Pharmaceutical ProductsMedhat Saad Eldin100% (1)
- Proscript User Manual Cms July 2014Document57 paginiProscript User Manual Cms July 2014Alia KhanÎncă nu există evaluări
- The Drug Discovery Process: Studies of Disease MechanismsDocument7 paginiThe Drug Discovery Process: Studies of Disease MechanismsSajanMaharjanÎncă nu există evaluări
- Week 3. DispensingDocument13 paginiWeek 3. DispensingDes LumabanÎncă nu există evaluări
- Prescription Verbs and Routes of AdminDocument2 paginiPrescription Verbs and Routes of AdminKingg AdjeiÎncă nu există evaluări
- Basic Concepts and Principles of PharmacokineticsDocument42 paginiBasic Concepts and Principles of PharmacokineticsSophia AgenyiÎncă nu există evaluări
- Essential of Pharmaceutical TechnologyDocument2 paginiEssential of Pharmaceutical TechnologyIbrahim Al SharabiÎncă nu există evaluări
- 14th Justa Causa BrochureDocument38 pagini14th Justa Causa BrochureAzharÎncă nu există evaluări
- Animal TestingDocument14 paginiAnimal TestingPer SheaÎncă nu există evaluări
- Study Guide and Sample Questions Exam 1 2018Document5 paginiStudy Guide and Sample Questions Exam 1 2018ParimalakrishnanÎncă nu există evaluări
- UGC Approved JournalsDocument22 paginiUGC Approved JournalsBMohdIshaqÎncă nu există evaluări
- Usp 2008 P 2 Supplement 3Document166 paginiUsp 2008 P 2 Supplement 3EstiPramestiningtyas100% (1)
- CROs in BelgiumDocument7 paginiCROs in BelgiumAna ConstantinescuÎncă nu există evaluări
- MARCH 23bills TALLY OUTSTANDING RECEIVABLE LIST 06.03.2023Document12 paginiMARCH 23bills TALLY OUTSTANDING RECEIVABLE LIST 06.03.2023radha gÎncă nu există evaluări
- Kaps GuideDocument3 paginiKaps Guideirnumambreen1550% (2)
- Drug Safety ReviewsDocument5 paginiDrug Safety ReviewsToronto StarÎncă nu există evaluări
- Gambaran Kejadian Medication Error Di Instalasi Gawat Darurat Rsu Elim RantepaoDocument7 paginiGambaran Kejadian Medication Error Di Instalasi Gawat Darurat Rsu Elim RantepaoRahmi Eka PutriÎncă nu există evaluări
- Rinvoq Extended Release Tablets 15 MGDocument8 paginiRinvoq Extended Release Tablets 15 MGMd Amdadul HoqueÎncă nu există evaluări
- 04 - Extravascular Administration (Oral)Document41 pagini04 - Extravascular Administration (Oral)Prasanna PappuÎncă nu există evaluări
- Pharmacology - PrelimsDocument9 paginiPharmacology - PrelimsLou KristofferÎncă nu există evaluări
- Biomedical Diagnostics Case Study Option 4Document6 paginiBiomedical Diagnostics Case Study Option 4Avril CoyleÎncă nu există evaluări
- Transcript: ـــــــــــــــــــــــــــــــــــــــــــ President ـــــــــــــــــــــــــــــــــــــــــــ RegistrarDocument1 paginăTranscript: ـــــــــــــــــــــــــــــــــــــــــــ President ـــــــــــــــــــــــــــــــــــــــــــ Registrarhamada234580Încă nu există evaluări
- Pdrugs and P TreatmentDocument64 paginiPdrugs and P TreatmentWendz BouvierÎncă nu există evaluări
- West Bengal Govt Health DistributorsDocument11 paginiWest Bengal Govt Health DistributorsAmit DesaiÎncă nu există evaluări