S-ar putea să vă placă și
- XRD 1 Lab 2018 10 01 PDFDocument9 paginiXRD 1 Lab 2018 10 01 PDF123hohoa123Încă nu există evaluări
- Infographic Analytical Tools For Decision MakersDocument10 paginiInfographic Analytical Tools For Decision Makerspedrovsky702Încă nu există evaluări
- Astroebsd: Exploring New Space in Pattern Indexing With Methods Launched From An Astronomical ApproachDocument10 paginiAstroebsd: Exploring New Space in Pattern Indexing With Methods Launched From An Astronomical ApproachLívia RodriguesÎncă nu există evaluări
- Does Your Lab Coat Fit To Your Assay?: Michael Busch, Heinz Bjoern Thoma, and Ingo KoberDocument4 paginiDoes Your Lab Coat Fit To Your Assay?: Michael Busch, Heinz Bjoern Thoma, and Ingo KoberkhosrosaneÎncă nu există evaluări
- Rietveld Refinement Practical Powder DiffractiDocument349 paginiRietveld Refinement Practical Powder Diffractixiao zhangÎncă nu există evaluări
- (Probability and Its Applications) Mu-Fa Chen - Eigenvalues, Inequalities, and Ergodic Theory (Probability and Its Applications) (2004, Springer) - Libgen - Li PDFDocument239 pagini(Probability and Its Applications) Mu-Fa Chen - Eigenvalues, Inequalities, and Ergodic Theory (Probability and Its Applications) (2004, Springer) - Libgen - Li PDFjkae romero100% (1)
- LAB3 Diffraction FT11Document7 paginiLAB3 Diffraction FT11Anushka RoyÎncă nu există evaluări
- N. Stribeck - X-Ray Scattering of Soft MatterDocument251 paginiN. Stribeck - X-Ray Scattering of Soft MatterElías Rodriguez100% (1)
- XRF ThesisDocument4 paginiXRF Thesisauroracuellarcostamesa100% (2)
- Cso202 Atomic and Molecular Beam MethodsDocument2 paginiCso202 Atomic and Molecular Beam MethodsAkhil ShuklaÎncă nu există evaluări
- Neumann, But Hold The Von: Articles You May Be Interested inDocument3 paginiNeumann, But Hold The Von: Articles You May Be Interested inDiego Alejandro Rasero CausilÎncă nu există evaluări
- Chemistry 843 "Advanced NMR Spectroscopy"Document6 paginiChemistry 843 "Advanced NMR Spectroscopy"luyawinÎncă nu există evaluări
- Handbook of Infrared Standards: With Spectral Maps and Transition Assignments Between 3 and 2600 x gmmDe la EverandHandbook of Infrared Standards: With Spectral Maps and Transition Assignments Between 3 and 2600 x gmmEvaluare: 1 din 5 stele1/5 (1)
- The First Periodic Table for Elementary ParticlesDe la EverandThe First Periodic Table for Elementary ParticlesÎncă nu există evaluări
- X Ray DiffractionDocument5 paginiX Ray DiffractionDeepak PatelÎncă nu există evaluări
- Differentiating Writing Inks Using Direct AnalysisDocument4 paginiDifferentiating Writing Inks Using Direct AnalysisIslam HelazaÎncă nu există evaluări
- Connoly 2005 Introduction To X-Ray Powder DiffractionDocument9 paginiConnoly 2005 Introduction To X-Ray Powder DiffractionFris BeeÎncă nu există evaluări
- Calculational Methods for Interacting Arrays of Fissile Material: International Series of Monographs in Nuclear EnergyDe la EverandCalculational Methods for Interacting Arrays of Fissile Material: International Series of Monographs in Nuclear EnergyÎncă nu există evaluări
- Experimental Methods and Instrumentation for Chemical EngineersDe la EverandExperimental Methods and Instrumentation for Chemical EngineersÎncă nu există evaluări
- Modern Polymer SpectroscopyDe la EverandModern Polymer SpectroscopyGiuseppe ZerbiÎncă nu există evaluări
- Reports On Progress in Physics Volume 24 Issue 1 1961 (Doi 10.1088/0034-4885/24/1/307) Haar, D Ter - Theory and Applications of The Density MatrixDocument60 paginiReports On Progress in Physics Volume 24 Issue 1 1961 (Doi 10.1088/0034-4885/24/1/307) Haar, D Ter - Theory and Applications of The Density MatrixDaniel BonillaÎncă nu există evaluări
- DFT PaperDocument12 paginiDFT PaperwpgurgelÎncă nu există evaluări
- Micro-nanoelectronics Devices: Modeling of Diffusion and Operation ProcessesDe la EverandMicro-nanoelectronics Devices: Modeling of Diffusion and Operation ProcessesÎncă nu există evaluări
- Title of The Manuscript in English Must Have Maximum 25 WordsDocument3 paginiTitle of The Manuscript in English Must Have Maximum 25 WordsLuis PaucarÎncă nu există evaluări
- Atom Probe Tomography: Put Theory Into PracticeDe la EverandAtom Probe Tomography: Put Theory Into PracticeWilliams LefebvreÎncă nu există evaluări
- Research Paper On XRDDocument7 paginiResearch Paper On XRDupvipbqlg100% (1)
- 01 XRD IntroDocument22 pagini01 XRD IntroSherlyn EnovejasÎncă nu există evaluări
- World's Largest ScienceDocument24 paginiWorld's Largest ScienceAnnisa Nur FitrianiÎncă nu există evaluări
- Nanomaterials Technical BulletinDocument16 paginiNanomaterials Technical BulletinIsmael SantosÎncă nu există evaluări
- Bayes Optimal Template Matching For Spike Sorting - Combining Fisher Discriminant Analysis With Optimal FilteringDocument21 paginiBayes Optimal Template Matching For Spike Sorting - Combining Fisher Discriminant Analysis With Optimal FilteringatewogboÎncă nu există evaluări
- (CMS Books in Mathematics 11) Bruce A. Reed, Claudia L. Linhares-Sales - Recent Advances in Algorithms and Combinatorics-Springer (2003)Document365 pagini(CMS Books in Mathematics 11) Bruce A. Reed, Claudia L. Linhares-Sales - Recent Advances in Algorithms and Combinatorics-Springer (2003)Chris AttongÎncă nu există evaluări
- XRD Standard Patterns of ZeoliteDocument586 paginiXRD Standard Patterns of ZeoliteVijaykumar MarakattiÎncă nu există evaluări
- Handbook of Infrared Standards II: with Spectral Coverage betweenDe la EverandHandbook of Infrared Standards II: with Spectral Coverage betweenÎncă nu există evaluări
- Error AnalysisDocument7 paginiError Analysiszpoturica569Încă nu există evaluări
- Methods of Radar Cross-section AnalysisDe la EverandMethods of Radar Cross-section AnalysisJ.W. Jr. CrispinÎncă nu există evaluări
- Seismic Amplitude Inversion in Reflection TomographyDe la EverandSeismic Amplitude Inversion in Reflection TomographyÎncă nu există evaluări
- Full-Field Measurements and Identification in Solid MechanicsDe la EverandFull-Field Measurements and Identification in Solid MechanicsMichel GrediacÎncă nu există evaluări
- Velten Research PaperDocument6 paginiVelten Research PaperkoshlendraÎncă nu există evaluări
- 5in-House Macromolecular Data CollectionDocument16 pagini5in-House Macromolecular Data CollectionAlfredoÎncă nu există evaluări
- Building A Spectroscope: Activity 1Document6 paginiBuilding A Spectroscope: Activity 1IVIYI3r41nÎncă nu există evaluări
- UNIT II Ingles IIDocument18 paginiUNIT II Ingles IIevelin gil larezÎncă nu există evaluări
- XRD HomeworkDocument5 paginiXRD Homeworkcfg1ngzn100% (1)
- Guided Waves in Structures for SHM: The Time - domain Spectral Element MethodDe la EverandGuided Waves in Structures for SHM: The Time - domain Spectral Element MethodÎncă nu există evaluări
- Group Theory and Its ApplicationsDe la EverandGroup Theory and Its ApplicationsErnest M. LoeblÎncă nu există evaluări
- Handbook of Sputter Deposition Technology: Fundamentals and Applications for Functional Thin Films, Nano-Materials and MEMSDe la EverandHandbook of Sputter Deposition Technology: Fundamentals and Applications for Functional Thin Films, Nano-Materials and MEMSEvaluare: 5 din 5 stele5/5 (1)
- Handbook of X-Ray SpectrometryDocument985 paginiHandbook of X-Ray Spectrometryjorgehrdz269100% (6)
- FH DissertationDocument8 paginiFH DissertationCanSomeoneWriteMyPaperRiverside100% (1)
- Prediction of Sheet Metal Formability (FLD) by Using Diverse MethodDocument11 paginiPrediction of Sheet Metal Formability (FLD) by Using Diverse Methodpraveen bavanaÎncă nu există evaluări
- Prediction of Sheet Metal Formability FLD by UsingDocument11 paginiPrediction of Sheet Metal Formability FLD by Usingpraveen bavanaÎncă nu există evaluări
- 318L TemplateDocument5 pagini318L Templatekurt2011Încă nu există evaluări
- MIT12 335F14 Lab1-ReportDocument4 paginiMIT12 335F14 Lab1-ReportMatthew VÎncă nu există evaluări
- Acta Numerica 1999 Volume 8 CompressDocument299 paginiActa Numerica 1999 Volume 8 CompressFernanda LuisaÎncă nu există evaluări
- Frac To GraphyDocument639 paginiFrac To GraphyBHARANIÎncă nu există evaluări
- Experiment-4: Aim: Synthesis and Characterization of Carbon Nanotube/Polypyrrole Requirements: Formula Used: TheoryDocument4 paginiExperiment-4: Aim: Synthesis and Characterization of Carbon Nanotube/Polypyrrole Requirements: Formula Used: TheoryTush RohÎncă nu există evaluări
- Tensile TestingDocument24 paginiTensile TestingMary TiltÎncă nu există evaluări
- 4CH1 2C Que 20211120Document24 pagini4CH1 2C Que 20211120Fazal AhmedÎncă nu există evaluări
- Design of Machine Elements 2019 BeemerDocument145 paginiDesign of Machine Elements 2019 BeemerSandeep MandaÎncă nu există evaluări
- Luxepoxy T: Tintable Two Pack Epoxy FinishDocument2 paginiLuxepoxy T: Tintable Two Pack Epoxy FinishlivefreakÎncă nu există evaluări
- Atoms and Molecules Reviewer 1 15Document5 paginiAtoms and Molecules Reviewer 1 15Vienna GilmoreÎncă nu există evaluări
- Medium Voltage Cables: Refineries & Petrochemical - OnshoreDocument2 paginiMedium Voltage Cables: Refineries & Petrochemical - OnshoreSriniÎncă nu există evaluări
- Crystex HD OT 20 PDFDocument2 paginiCrystex HD OT 20 PDFmeidyÎncă nu există evaluări
- Crystal Field Theory IIDocument2 paginiCrystal Field Theory IIabhay j bavishiÎncă nu există evaluări
- Geochemical Analysis of Iron Ore - SGSDocument2 paginiGeochemical Analysis of Iron Ore - SGSAnton De la ruaÎncă nu există evaluări
- Used Oil Recycling and Treatment in The United AraDocument11 paginiUsed Oil Recycling and Treatment in The United AraEssam AlharthyÎncă nu există evaluări
- HVAC Water TreatmentDocument6 paginiHVAC Water TreatmentAbdullah.N FAAliÎncă nu există evaluări
- Nanomaterials in Structural EngineeringDocument19 paginiNanomaterials in Structural EngineeringAniket DubeÎncă nu există evaluări
- Methodology For Petrophysical and Geomechanical Analysis of Shale Plays. Study Case: La Luna and Capacho Formations, Maracaibo Basin. Presentation of Paper SPE-185606-MSDocument24 paginiMethodology For Petrophysical and Geomechanical Analysis of Shale Plays. Study Case: La Luna and Capacho Formations, Maracaibo Basin. Presentation of Paper SPE-185606-MSCarlos LoboÎncă nu există evaluări
- CAPE Unit 2 LabsDocument4 paginiCAPE Unit 2 LabsAlex Clarke50% (6)
- Ultra-Violet Coatings Tricks and TipsDocument1 paginăUltra-Violet Coatings Tricks and TipsAdriano AraujoÎncă nu există evaluări
- Pacing Guide: Inspire ChemistryDocument6 paginiPacing Guide: Inspire Chemistryjsencion977Încă nu există evaluări
- Basics of Paint TechnologyDocument9 paginiBasics of Paint TechnologySantosh Raj100% (1)
- Chlodnice Oleju CSL CiesseDocument28 paginiChlodnice Oleju CSL CiesseCARLOS RAMIREZÎncă nu există evaluări
- Welding of Duplex Stainless SteelDocument7 paginiWelding of Duplex Stainless Steelel_sharkawy2011Încă nu există evaluări
- Extracting MetalsDocument27 paginiExtracting MetalsMadan Yadav100% (2)
- Stefan Boltzmann Law PDFDocument3 paginiStefan Boltzmann Law PDFESAKKIMALA SÎncă nu există evaluări
- MSDS Burnshield Sachets 4BDocument5 paginiMSDS Burnshield Sachets 4BJarrod Currin100% (1)
- Nanoformulation of Curcuma Longa Root Extract and Evaluation of Its Dissolution PotentialDocument9 paginiNanoformulation of Curcuma Longa Root Extract and Evaluation of Its Dissolution Potentialmuhammad adnan ayubÎncă nu există evaluări
- TDS-Dow SPECFIL FT630 & SPECFIL FE100-EN - 20181226Document2 paginiTDS-Dow SPECFIL FT630 & SPECFIL FE100-EN - 20181226Mallampati RamakrishnaÎncă nu există evaluări
- Protego 2020 - 21Document429 paginiProtego 2020 - 21jleonclau1Încă nu există evaluări
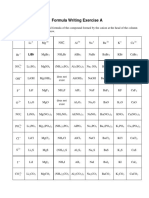
- Formula Writing - CambridgeDocument5 paginiFormula Writing - CambridgeQusai Saify100% (3)
- DPP 9Document3 paginiDPP 9Sarvesh DubeyÎncă nu există evaluări
- ElectrophoresisDocument47 paginiElectrophoresisEllah GutierrezÎncă nu există evaluări
- 30 Sewage Treatment PlanDocument63 pagini30 Sewage Treatment PlanYuri Duri100% (1)
- FORMULATION AND EVALUATION OF GASTRO-RETENTIVE FLOATING TABLET OF QUETIAPINE FUMARATE Shanti Sagar, Srividya. L, B.K NanjawadeDocument14 paginiFORMULATION AND EVALUATION OF GASTRO-RETENTIVE FLOATING TABLET OF QUETIAPINE FUMARATE Shanti Sagar, Srividya. L, B.K NanjawadeiajpsÎncă nu există evaluări