S-ar putea să vă placă și
- Selecting The Right' Positive End-Expiratory Pressure Level (Luciano Gattinoni, Current Opinion, 2016)Document8 paginiSelecting The Right' Positive End-Expiratory Pressure Level (Luciano Gattinoni, Current Opinion, 2016)PkernÎncă nu există evaluări
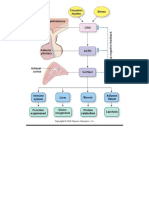
- Simple Adrenal Synthesis PathwayDocument1 paginăSimple Adrenal Synthesis PathwayPkernÎncă nu există evaluări
- 6th Central Pay Commission Salary CalculatorDocument15 pagini6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Surgical Procedures in The Intensive Care Unit - A Critical Review. (BM Dennis, OL Gunter OA CC 2013)Document7 paginiSurgical Procedures in The Intensive Care Unit - A Critical Review. (BM Dennis, OL Gunter OA CC 2013)PkernÎncă nu există evaluări
- The Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFDocument10 paginiThe Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFPkernÎncă nu există evaluări
- The Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFDocument10 paginiThe Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFPkernÎncă nu există evaluări
- The Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFDocument10 paginiThe Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFPkernÎncă nu există evaluări
- The Crashing Ventilated Patient Algorthm (Jairo I. Santanilla, ACEP, 2011)Document1 paginăThe Crashing Ventilated Patient Algorthm (Jairo I. Santanilla, ACEP, 2011)PkernÎncă nu există evaluări
- Diuretics and Renal HormonesDocument3 paginiDiuretics and Renal HormonesPkernÎncă nu există evaluări
- Primary FRCA Pharmacology EquationsDocument11 paginiPrimary FRCA Pharmacology EquationsMustafa SharkawyÎncă nu există evaluări
- Rule of 4 Illustration Brainstem (LITFL)Document1 paginăRule of 4 Illustration Brainstem (LITFL)PkernÎncă nu există evaluări
- Primary FRCA Pharmacology EquationsDocument11 paginiPrimary FRCA Pharmacology EquationsMustafa SharkawyÎncă nu există evaluări
- Heart Failure PDFDocument9 paginiHeart Failure PDFPkernÎncă nu există evaluări
- Spinning Dials - How To Dominate The Ventilator - Handout (EMCRIT, ARDSnet) PDFDocument5 paginiSpinning Dials - How To Dominate The Ventilator - Handout (EMCRIT, ARDSnet) PDFPkernÎncă nu există evaluări
- Respiratory-Equations (Adam Hollingworth)Document4 paginiRespiratory-Equations (Adam Hollingworth)PkernÎncă nu există evaluări
- Human Metabolism MapDocument1 paginăHuman Metabolism MapPkern83% (6)
- STRUCTURE AND FUNCTION OF THE HEARTDocument16 paginiSTRUCTURE AND FUNCTION OF THE HEARTPkernÎncă nu există evaluări
- Anaesthetic Drugs Cheat SheetsDocument2 paginiAnaesthetic Drugs Cheat SheetsPkern100% (3)
- Emergency Department Drug Card - For ALIEM - Sep 2013Document1 paginăEmergency Department Drug Card - For ALIEM - Sep 2013PkernÎncă nu există evaluări
- ED Drug Treatment Cheat Sheet ANZICSDocument1 paginăED Drug Treatment Cheat Sheet ANZICSPkernÎncă nu există evaluări
- Chapter 3 The Lymphatic VasculatureDocument8 paginiChapter 3 The Lymphatic VasculaturePkernÎncă nu există evaluări
- Renal, Fluids and Acid-Base Notes (Chris Andersen, ICUPrimaryPrep - Com)Document8 paginiRenal, Fluids and Acid-Base Notes (Chris Andersen, ICUPrimaryPrep - Com)PkernÎncă nu există evaluări
- Neurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Document11 paginiNeurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Pkern100% (1)
- Cardiac Murmurs and Maneuvers GuideDocument1 paginăCardiac Murmurs and Maneuvers GuidePkernÎncă nu există evaluări
- Pharmacodynamics and Receptor TheoryDocument8 paginiPharmacodynamics and Receptor TheoryPkernÎncă nu există evaluări
- Neurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Document11 paginiNeurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Pkern100% (1)
- Chapter 4 Pathophysiology of Edema FormationDocument8 paginiChapter 4 Pathophysiology of Edema FormationPkernÎncă nu există evaluări
- 2014 CCM Review Notes Jon-Emile S. Kenny M.D, 2014Document142 pagini2014 CCM Review Notes Jon-Emile S. Kenny M.D, 2014PkernÎncă nu există evaluări
- Respiratory Notes (Chris Andersen, ICUPrimaryPrep - Com)Document14 paginiRespiratory Notes (Chris Andersen, ICUPrimaryPrep - Com)PkernÎncă nu există evaluări
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5784)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (399)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (890)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (587)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (265)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (72)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (119)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- Acute Limb Ischemia and Compartment Syndrome OverviewDocument21 paginiAcute Limb Ischemia and Compartment Syndrome OverviewHamzeh K. YacoubÎncă nu există evaluări
- 2.02 Gross Anatomy Trans - HeartDocument15 pagini2.02 Gross Anatomy Trans - HeartElma Gonzales100% (1)
- A Pocket Guide To Blood Pressure Measurement in ChildrenDocument4 paginiA Pocket Guide To Blood Pressure Measurement in ChildrenKira23406Încă nu există evaluări
- Final PresentationDocument25 paginiFinal Presentationapi-520229867Încă nu există evaluări
- ECG MachinesDocument36 paginiECG Machinesrobert robÎncă nu există evaluări
- Basic Life Support (BLS) : Franklin O. Que, BSN-RNDocument90 paginiBasic Life Support (BLS) : Franklin O. Que, BSN-RNfrank_1785Încă nu există evaluări
- Dr. Witra Irfan, SP.B (K) V - DVTDocument29 paginiDr. Witra Irfan, SP.B (K) V - DVTmuhammad azharanÎncă nu există evaluări
- Circ TESTDocument5 paginiCirc TESTCabo VlogÎncă nu există evaluări
- Lymphatic System ReportDocument6 paginiLymphatic System ReportcrtgyhujikÎncă nu există evaluări
- Radiol 223008Document15 paginiRadiol 223008rehan hayderÎncă nu există evaluări
- Reference 2Document4 paginiReference 2Tari De ArimbieÎncă nu există evaluări
- Cardiogenic Shock NCLEX Review QuizDocument6 paginiCardiogenic Shock NCLEX Review QuizRegie Marie EvangelistaÎncă nu există evaluări
- Angina Pectoris: Akshay AgrawalDocument22 paginiAngina Pectoris: Akshay AgrawalBheru LalÎncă nu există evaluări
- Brochure - MagicTouch, Concept MedicalDocument6 paginiBrochure - MagicTouch, Concept MedicalBUSHRA ILYASÎncă nu există evaluări
- Aortic RegurgitationDocument41 paginiAortic RegurgitationMedi PutraÎncă nu există evaluări
- EsmololDocument2 paginiEsmololtherock316_995149Încă nu există evaluări
- Rheumatic Fever and Rheumatic Heart DiseaseDocument54 paginiRheumatic Fever and Rheumatic Heart DiseaseFian AldyÎncă nu există evaluări
- Heart Sounds Practical GuideDocument12 paginiHeart Sounds Practical GuideDratosh KatiyarÎncă nu există evaluări
- Assessment of Vital Signs&GCSDocument3 paginiAssessment of Vital Signs&GCSNicole Jackson100% (3)
- Neonatal Transition: Training of The Trainers Neonatal ResuscitationDocument36 paginiNeonatal Transition: Training of The Trainers Neonatal ResuscitationmitaÎncă nu există evaluări
- Focal Atrial Tachycardia I: Clinical Features, DiagnosisDocument10 paginiFocal Atrial Tachycardia I: Clinical Features, Diagnosisapi-26166949Încă nu există evaluări
- Virchow TriadDocument6 paginiVirchow Triadarif 2006Încă nu există evaluări
- Kasus Lain LainDocument14 paginiKasus Lain LainofislinÎncă nu există evaluări
- PIT XI Perhimpunan Ahli Bedah Toraks, Kardiak Dan Vaskular Indonesia (HBTKVI)Document1 paginăPIT XI Perhimpunan Ahli Bedah Toraks, Kardiak Dan Vaskular Indonesia (HBTKVI)RakaÎncă nu există evaluări
- Ecg - Partea I - Mircea CintezaDocument16 paginiEcg - Partea I - Mircea CintezaAndrei CiobotaruÎncă nu există evaluări
- ACS Case StudyDocument26 paginiACS Case StudyMari Lyn100% (1)
- Venipuncture Module 1 - Anatomy of The Arm and Vein LocationDocument21 paginiVenipuncture Module 1 - Anatomy of The Arm and Vein LocationAulita AlphonceÎncă nu există evaluări
- Perfusion SystemDocument52 paginiPerfusion Systemprafilaash100% (1)
- Cardiology SoalDocument9 paginiCardiology Soalanz_4191Încă nu există evaluări
- Cor Pulmonale - StatPearls - NCBI BookshelfDocument4 paginiCor Pulmonale - StatPearls - NCBI BookshelfAldi RafaelÎncă nu există evaluări