S-ar putea să vă placă și
- Cancer and The CellDocument10 paginiCancer and The CellaxlzekeÎncă nu există evaluări
- Group3 Errors in Cell CycleDocument26 paginiGroup3 Errors in Cell CycleRhian CodilanÎncă nu există evaluări
- Cancer GeneticsDocument11 paginiCancer GeneticsFuture TrekingÎncă nu există evaluări
- Regulating The Cell Cycle: Lesson SummaryDocument7 paginiRegulating The Cell Cycle: Lesson SummaryKayÎncă nu există evaluări
- What Are The Biochemical and Structural Changes That Occur in Cancer CellDocument7 paginiWhat Are The Biochemical and Structural Changes That Occur in Cancer CellsujsamÎncă nu există evaluări
- Cancer (Medicine), Any of More Than 100 Diseases Characterized by ExcessiveDocument30 paginiCancer (Medicine), Any of More Than 100 Diseases Characterized by ExcessiveKwaku Essilfie100% (1)
- Principles & CancerDocument28 paginiPrinciples & CancerAna Abuladze100% (1)
- Replicative SenescenceDocument3 paginiReplicative SenescenceJasmine Bernadette CubillaÎncă nu există evaluări
- Introduction to Cellular Aberration: Normal Cell vs. Cancer CellDocument11 paginiIntroduction to Cellular Aberration: Normal Cell vs. Cancer CellAdiel CalsaÎncă nu există evaluări
- Cancer - Zarah DDocument3 paginiCancer - Zarah DzarahsabinÎncă nu există evaluări
- LESSON 3 Cell CycleDocument10 paginiLESSON 3 Cell CycleAiza CeciliaÎncă nu există evaluări
- Healthy You Processed Foods Wk. 4Document49 paginiHealthy You Processed Foods Wk. 4Nathaly DiazÎncă nu există evaluări
- Cell Division Regulation and Cancer GrowthDocument7 paginiCell Division Regulation and Cancer GrowthFRISKA CHRISTININGRUMÎncă nu există evaluări
- GB1 - W4 Module 2 1st QTR 1Document145 paginiGB1 - W4 Module 2 1st QTR 1AyaÎncă nu există evaluări
- Biology of CancerDocument59 paginiBiology of CancerMhmoud NabehÎncă nu există evaluări
- Assignment # 01Document3 paginiAssignment # 01Daniyal ArifÎncă nu există evaluări
- The Eight Hallmarks of CancerDocument6 paginiThe Eight Hallmarks of Cancercaraphernelia ;Încă nu există evaluări
- Saima Victor KAH College of Health Sciences 1Document8 paginiSaima Victor KAH College of Health Sciences 1Saima VictorÎncă nu există evaluări
- Oncolgy UnitDocument12 paginiOncolgy UnitAtia IftikharÎncă nu există evaluări
- Cancer Cell Development - Lecture Day 5Document17 paginiCancer Cell Development - Lecture Day 5blakeÎncă nu există evaluări
- Cancer Cells: How They Start and CharacteristicsDocument7 paginiCancer Cells: How They Start and CharacteristicsARISÎncă nu există evaluări
- Thursday, 3 November 2022 10:14 PMDocument5 paginiThursday, 3 November 2022 10:14 PMAdiel CalsaÎncă nu există evaluări
- Biology Project OncolgyDocument23 paginiBiology Project OncolgyAyaan Raza KhanÎncă nu există evaluări
- 9 3Document23 pagini9 3nancie8Încă nu există evaluări
- What Is Cancer Cellhow Does It DevelopDocument3 paginiWhat Is Cancer Cellhow Does It DevelopStephanie Nichole Ian CasemÎncă nu există evaluări
- Breast Cancer Pathophysiology Cancer PathophysiologyDocument4 paginiBreast Cancer Pathophysiology Cancer PathophysiologyprathibaÎncă nu există evaluări
- Module 2 Proliferative Growth Patterns Etiology Role of The Immune SystemDocument69 paginiModule 2 Proliferative Growth Patterns Etiology Role of The Immune SystemAlessandra MercadoÎncă nu există evaluări
- CCHM 312: Week 7: Tumor MarkersDocument5 paginiCCHM 312: Week 7: Tumor MarkersJanine Alicia VargasÎncă nu există evaluări
- Cancer: Cancer Is Defined As An Uncontrolled Proliferation ofDocument12 paginiCancer: Cancer Is Defined As An Uncontrolled Proliferation ofnikÎncă nu există evaluări
- Nursing OncologyDocument124 paginiNursing OncologyAmisalu NigusieÎncă nu există evaluări
- Module 1: Pathophysiologic Concepts in Cellular AberrationsDocument9 paginiModule 1: Pathophysiologic Concepts in Cellular AberrationsJanelle Cabida SupnadÎncă nu există evaluări
- The Hallmarks of CancerDocument7 paginiThe Hallmarks of Cancerari purnamayasaÎncă nu există evaluări
- Cells: The Building Blocks of LifeDocument2 paginiCells: The Building Blocks of LifemimiÎncă nu există evaluări
- Reviewer - Cellular AberrationDocument26 paginiReviewer - Cellular AberrationRaquel HatulanÎncă nu există evaluări
- What Is CancersDocument17 paginiWhat Is Cancersmalaran.deboraheloisaÎncă nu există evaluări
- Explain Why Cancer Is Regarded As Abnormal Cellular FunctionDocument1 paginăExplain Why Cancer Is Regarded As Abnormal Cellular FunctionCJ PrettyÎncă nu există evaluări
- Aging Guides: Cellular SenescenceDocument6 paginiAging Guides: Cellular SenescenceKana FajarÎncă nu există evaluări
- Articles Bu Ke 1Document3 paginiArticles Bu Ke 1dina aribahÎncă nu există evaluări
- B 10 VRV 3103Document14 paginiB 10 VRV 3103api-283593849Încă nu există evaluări
- Onco, TSG & CancerDocument8 paginiOnco, TSG & Cancersumera120488Încă nu există evaluări
- Cell Cycle Regulation and CheckpointsDocument3 paginiCell Cycle Regulation and Checkpointsʀᴇɢᴇʟʏɴ ʙᴀʟɪᴅÎncă nu există evaluări
- Cancer Types, Causes, and DevelopmentDocument1 paginăCancer Types, Causes, and DevelopmentrajeshmangalÎncă nu există evaluări
- ! Cellular and Epigenetic Drivers of Stem Cell Ageing 2018 Review PDFDocument17 pagini! Cellular and Epigenetic Drivers of Stem Cell Ageing 2018 Review PDF畏Încă nu există evaluări
- Tumor Suppressor OnkogenDocument4 paginiTumor Suppressor OnkogenFlorentina Lisa PratamaÎncă nu există evaluări
- How Cancer Cells Grow and SpreadDocument1 paginăHow Cancer Cells Grow and SpreadFatima SantosÎncă nu există evaluări
- The CEllular AberrationsDocument17 paginiThe CEllular AberrationsShara Sampang100% (1)
- MSU Marawi Cellular Aberration Final Exam AnswersDocument3 paginiMSU Marawi Cellular Aberration Final Exam AnswersSajeebChandraÎncă nu există evaluări
- Hallmarks of CancerDocument2 paginiHallmarks of CancerHarvey Lampa SelimÎncă nu există evaluări
- Cell Cycle (Rubina)Document35 paginiCell Cycle (Rubina)RubinaÎncă nu există evaluări
- Cellular AberrationDocument6 paginiCellular AberrationLance RaphaelÎncă nu există evaluări
- Case Study: Breast CancerDocument24 paginiCase Study: Breast CancerCutie KidsArtÎncă nu există evaluări
- The Story of Our Cells: Cancer, Growth, Mitosis, Importance in 40 CharactersDocument8 paginiThe Story of Our Cells: Cancer, Growth, Mitosis, Importance in 40 CharactersDanKimberleyÎncă nu există evaluări
- Cancer Vs Normal Cell1111Document25 paginiCancer Vs Normal Cell1111JarrarÎncă nu există evaluări
- Week 3 Modules Grade 12Document13 paginiWeek 3 Modules Grade 12Joe NasalitaÎncă nu există evaluări
- Cellular AberrationDocument10 paginiCellular AberrationBiway RegalaÎncă nu există evaluări
- What Causes Cancer and How It DevelopsDocument30 paginiWhat Causes Cancer and How It DevelopsagengbsÎncă nu există evaluări
- 1 Definition of Cancers 1 4Document4 pagini1 Definition of Cancers 1 4nikiÎncă nu există evaluări
- 14 - PoleñoDocument9 pagini14 - PoleñoPebbles PangilinanÎncă nu există evaluări
- Writing The IntroductionDocument3 paginiWriting The IntroductionjayweinxÎncă nu există evaluări
- It Must Be Halloween: 1. DiscussDocument5 paginiIt Must Be Halloween: 1. DiscussjayweinxÎncă nu există evaluări
- Introduction to Biology and Laboratory RulesDocument142 paginiIntroduction to Biology and Laboratory RulesRouXuanChangÎncă nu există evaluări
- Talking About Money: 1. DiscussDocument4 paginiTalking About Money: 1. Discussjayweinx100% (1)
- The Scene at A Crowded Shopping MallDocument4 paginiThe Scene at A Crowded Shopping MalljayweinxÎncă nu există evaluări
- CW Compilation (2017 Trials)Document4 paginiCW Compilation (2017 Trials)FBÎncă nu există evaluări
- How A Typical Five-Paragraph Essay Is Written SCDocument4 paginiHow A Typical Five-Paragraph Essay Is Written SCjayweinxÎncă nu există evaluări
- My HeroDocument7 paginiMy HerojayweinxÎncă nu există evaluări
- Novel: King Arthur: Theme: courage 勇敢Document3 paginiNovel: King Arthur: Theme: courage 勇敢jayweinxÎncă nu există evaluări
- Mid Year f2 2016Document14 paginiMid Year f2 2016jayweinxÎncă nu există evaluări
- f5 110319Document3 paginif5 110319jayweinxÎncă nu există evaluări
- January 1Document49 paginiJanuary 1LAMCÎncă nu există evaluări
- 体面Document1 pagină体面jayweinxÎncă nu există evaluări
- Pep. Set 4 PT3 2016 PDFDocument2 paginiPep. Set 4 PT3 2016 PDFjayweinx100% (1)
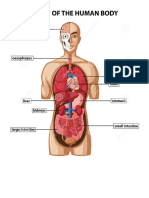
- Anatomy of Human BodyDocument1 paginăAnatomy of Human BodyjayweinxÎncă nu există evaluări
- Report On Co-CurricularDocument3 paginiReport On Co-CurricularjayweinxÎncă nu există evaluări
- Directed Writing Report About The School CanteenDocument4 paginiDirected Writing Report About The School Canteenjayweinx100% (2)
- Chanyeol EXO Punch - Stay With MeDocument3 paginiChanyeol EXO Punch - Stay With MejayweinxÎncă nu există evaluări
- 5 CBA HEART ATTACK GUIDEDocument5 pagini5 CBA HEART ATTACK GUIDEjayweinxÎncă nu există evaluări
- Ibmyp2 10Document11 paginiIbmyp2 10jayweinxÎncă nu există evaluări
- Anatomy of Human BodyDocument1 paginăAnatomy of Human BodyjayweinxÎncă nu există evaluări
- Ujian Sumatif 1 Ting 1 2017Document8 paginiUjian Sumatif 1 Ting 1 2017jayweinxÎncă nu există evaluări
- Line EeeeDocument1 paginăLine EeeejayweinxÎncă nu există evaluări
- Algebraic Expressions ExerciseDocument18 paginiAlgebraic Expressions ExerciseLi Ann ChungÎncă nu există evaluări
- Modul 2 PDF July 13 2009 9 39 PM 181kDocument10 paginiModul 2 PDF July 13 2009 9 39 PM 181kArun ManiamÎncă nu există evaluări
- English Test Written Test GrammarDocument10 paginiEnglish Test Written Test Grammartarubi53100% (3)
- Tips 2017 SPM EngDocument13 paginiTips 2017 SPM EngjayweinxÎncă nu există evaluări
- Here are the algebraic expressions for the given statements:2 more than x = x + 26 less than x = x - 6 k more than x = x + kx minus t = x - tx added t = x + tDocument52 paginiHere are the algebraic expressions for the given statements:2 more than x = x + 26 less than x = x - 6 k more than x = x + kx minus t = x - tx added t = x + tjayweinxÎncă nu există evaluări
- C1 Maths F1 Topical Test 1Document3 paginiC1 Maths F1 Topical Test 1jayweinxÎncă nu există evaluări
- f3 Bi Conjuction, Application LetterDocument7 paginif3 Bi Conjuction, Application LetterjayweinxÎncă nu există evaluări
- Test Bank For Molecular Biology 5th Edition Robert WeaverDocument36 paginiTest Bank For Molecular Biology 5th Edition Robert Weaverbrokeazarole6rv8pl100% (43)
- Genetic and Biological Hallmarks of Colorectal CancerDocument34 paginiGenetic and Biological Hallmarks of Colorectal CancerFredÎncă nu există evaluări
- STEM - Biology 2 CG - With Tagged Sci EquipmentDocument4 paginiSTEM - Biology 2 CG - With Tagged Sci EquipmentVictoria MabiniÎncă nu există evaluări
- (PDF) GenBio2 - Module - Week06Document40 pagini(PDF) GenBio2 - Module - Week06Ramon Villota100% (1)
- Chapter 1.1Document70 paginiChapter 1.1AS2012B2 FATIN NUR AQILAH BINTI DAZALIÎncă nu există evaluări
- Janet RowleyDocument1 paginăJanet RowleyANA GABRIELA PEREZ VIDALÎncă nu există evaluări
- 18 DNA Structure and Replication-SDocument5 pagini18 DNA Structure and Replication-Sjerhebeh100% (2)
- Prader Willi SyndromeDocument14 paginiPrader Willi Syndromeapi-471834071Încă nu există evaluări
- Lab 7 ReportDocument9 paginiLab 7 ReportZach ThorpeÎncă nu există evaluări
- THE PHYSICAL SELF: STAYING FIT THROUGH DIET, SLEEP AND EXERCISEDocument9 paginiTHE PHYSICAL SELF: STAYING FIT THROUGH DIET, SLEEP AND EXERCISENoelyn PaghubasanÎncă nu există evaluări
- Biology and Evolution of Life ScienceDocument5 paginiBiology and Evolution of Life ScienceAdissa Hasna MutiaraÎncă nu există evaluări
- Is Homosexuality Genetic?Document3 paginiIs Homosexuality Genetic?Larissa ReateguiÎncă nu există evaluări
- Newsletter: Plant Mutation BreedingDocument3 paginiNewsletter: Plant Mutation BreedingGabbyÎncă nu există evaluări
- Optimal Project Planning For Public Rental Housing in South KoreaDocument14 paginiOptimal Project Planning For Public Rental Housing in South Korearuth agittaÎncă nu există evaluări
- 4transf MaizDocument13 pagini4transf MaizIris MoralesÎncă nu există evaluări
- 05pop EcologyDocument5 pagini05pop Ecologynoor nadhirahÎncă nu există evaluări
- Genetics - HumanKaryotyping - GizmoDocument4 paginiGenetics - HumanKaryotyping - GizmoRicky DunlapÎncă nu există evaluări
- WORKSHEET 14.3 Sex Determination in OffspringDocument3 paginiWORKSHEET 14.3 Sex Determination in OffspringafzalÎncă nu există evaluări
- Views of Orthodox Theology on Bioethical IssuesDocument24 paginiViews of Orthodox Theology on Bioethical IssuessilviustoiangalatiÎncă nu există evaluări
- Amoeba Sisters Meiosis Video RecapDocument3 paginiAmoeba Sisters Meiosis Video RecapLESLEY AGUIRREÎncă nu există evaluări
- Isolation and Purification of Putative Pentr4:Campari ClonesDocument10 paginiIsolation and Purification of Putative Pentr4:Campari ClonesBrittani HyltonÎncă nu există evaluări
- Gene Testing: Naresh ButaniDocument17 paginiGene Testing: Naresh ButaniNaresh ButaniÎncă nu există evaluări
- Dhiraj Kumar, Chengliang Gong-Trends in Insect Molecular Biology and Biotechnology-Springer International Publishing (2018) PDFDocument376 paginiDhiraj Kumar, Chengliang Gong-Trends in Insect Molecular Biology and Biotechnology-Springer International Publishing (2018) PDFAndres Felipe Arias MosqueraÎncă nu există evaluări
- Usama Sumalani Biology NotesDocument3 paginiUsama Sumalani Biology NotesXë ShãñÎncă nu există evaluări
- Students Will Be Called Randomly.Document5 paginiStudents Will Be Called Randomly.Naisy Magalona100% (1)
- NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models Via Mitophagy and DNA RepairDocument47 paginiNAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models Via Mitophagy and DNA Repairender000100% (1)
- SAADC2015 Abstract BookDocument372 paginiSAADC2015 Abstract Bookfiqi iqbalÎncă nu există evaluări
- Lesson 1 Cellular Processes PPTDocument19 paginiLesson 1 Cellular Processes PPTkj47ynpdh7Încă nu există evaluări
- Heredity and EvolutionDocument5 paginiHeredity and EvolutionRahul Ecka100% (3)
- Sci and Tech ReadingsDocument26 paginiSci and Tech Readingsdaniel_siewÎncă nu există evaluări
- 10% Human: How Your Body's Microbes Hold the Key to Health and HappinessDe la Everand10% Human: How Your Body's Microbes Hold the Key to Health and HappinessEvaluare: 4 din 5 stele4/5 (33)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisDe la EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisEvaluare: 3.5 din 5 stele3.5/5 (2)
- Why We Die: The New Science of Aging and the Quest for ImmortalityDe la EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityEvaluare: 3.5 din 5 stele3.5/5 (2)
- Crypt: Life, Death and Disease in the Middle Ages and BeyondDe la EverandCrypt: Life, Death and Disease in the Middle Ages and BeyondEvaluare: 4 din 5 stele4/5 (3)
- This Is Your Brain On Parasites: How Tiny Creatures Manipulate Our Behavior and Shape SocietyDe la EverandThis Is Your Brain On Parasites: How Tiny Creatures Manipulate Our Behavior and Shape SocietyEvaluare: 3.5 din 5 stele3.5/5 (31)
- The Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionDe la EverandThe Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionEvaluare: 4 din 5 stele4/5 (811)
- Masterminds: Genius, DNA, and the Quest to Rewrite LifeDe la EverandMasterminds: Genius, DNA, and the Quest to Rewrite LifeÎncă nu există evaluări
- The Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindDe la EverandThe Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindEvaluare: 4.5 din 5 stele4.5/5 (93)
- The Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorDe la EverandThe Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorÎncă nu există evaluări
- All That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesDe la EverandAll That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesEvaluare: 4.5 din 5 stele4.5/5 (397)
- The Lives of Bees: The Untold Story of the Honey Bee in the WildDe la EverandThe Lives of Bees: The Untold Story of the Honey Bee in the WildEvaluare: 4.5 din 5 stele4.5/5 (44)
- Undeniable: How Biology Confirms Our Intuition That Life Is DesignedDe la EverandUndeniable: How Biology Confirms Our Intuition That Life Is DesignedEvaluare: 4 din 5 stele4/5 (11)
- The Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceDe la EverandThe Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceEvaluare: 4.5 din 5 stele4.5/5 (515)
- Human Errors: A Panorama of Our Glitches, from Pointless Bones to Broken GenesDe la EverandHuman Errors: A Panorama of Our Glitches, from Pointless Bones to Broken GenesEvaluare: 3.5 din 5 stele3.5/5 (56)
- Wayfinding: The Science and Mystery of How Humans Navigate the WorldDe la EverandWayfinding: The Science and Mystery of How Humans Navigate the WorldEvaluare: 4.5 din 5 stele4.5/5 (18)
- The Mind & The Brain: Neuroplasticity and the Power of Mental ForceDe la EverandThe Mind & The Brain: Neuroplasticity and the Power of Mental ForceÎncă nu există evaluări
- A Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsDe la EverandA Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsEvaluare: 4.5 din 5 stele4.5/5 (4)
- Eels: An Exploration, from New Zealand to the Sargasso, of the World's Most Mysterious FishDe la EverandEels: An Exploration, from New Zealand to the Sargasso, of the World's Most Mysterious FishEvaluare: 4 din 5 stele4/5 (30)
- Gathering Moss: A Natural and Cultural History of MossesDe la EverandGathering Moss: A Natural and Cultural History of MossesEvaluare: 4.5 din 5 stele4.5/5 (347)
- The Second Brain: A Groundbreaking New Understanding of Nervous Disorders of the Stomach and IntestineDe la EverandThe Second Brain: A Groundbreaking New Understanding of Nervous Disorders of the Stomach and IntestineEvaluare: 4 din 5 stele4/5 (17)
- Darwin's Dangerous Idea: Evolution and the Meaning of LifeDe la EverandDarwin's Dangerous Idea: Evolution and the Meaning of LifeEvaluare: 4 din 5 stele4/5 (523)
- Younger for Life: Feel Great and Look Your Best with the New Science of AutojuvenationDe la EverandYounger for Life: Feel Great and Look Your Best with the New Science of AutojuvenationEvaluare: 4 din 5 stele4/5 (1)
- Inside of a Dog: What Dogs See, Smell, and KnowDe la EverandInside of a Dog: What Dogs See, Smell, and KnowEvaluare: 4 din 5 stele4/5 (390)
- Superlative: The Biology of ExtremesDe la EverandSuperlative: The Biology of ExtremesEvaluare: 4.5 din 5 stele4.5/5 (51)
- The Magic of Reality: How We Know What's Really TrueDe la EverandThe Magic of Reality: How We Know What's Really TrueEvaluare: 4.5 din 5 stele4.5/5 (106)
- Lymph & Longevity: The Untapped Secret to HealthDe la EverandLymph & Longevity: The Untapped Secret to HealthEvaluare: 4.5 din 5 stele4.5/5 (13)