S-ar putea să vă placă și
- Centrómeros y TelomerosDocument4 paginiCentrómeros y TelomerosRicardoÎncă nu există evaluări
- Aplicaciones Educativas Dispositivos MovilesDocument7 paginiAplicaciones Educativas Dispositivos Movilesyamireth_16Încă nu există evaluări
- Diseño Instruccional y Materiales DidacticosDocument12 paginiDiseño Instruccional y Materiales DidacticosJuan Gabriel Moreno LeónÎncă nu există evaluări
- Plan de MKT Industria GraficaDocument7 paginiPlan de MKT Industria GraficaDILBHÎncă nu există evaluări
- Mi Experiencia en Second LifeDocument3 paginiMi Experiencia en Second LifeAngel Oswaldo Romero HinojozaÎncă nu există evaluări
- Glandulas MamariasDocument4 paginiGlandulas MamariasYelitza GarceteÎncă nu există evaluări
- Reacciones AdversasDocument15 paginiReacciones AdversasWaleska Marín MercadoÎncă nu există evaluări
- Las Innovaciones Educativas en La Profesionalizacion DocenteDocument13 paginiLas Innovaciones Educativas en La Profesionalizacion DocenteNellys CastilloÎncă nu există evaluări
- TIM, Matriz de Integración de TIC en Procesos EducativosDocument6 paginiTIM, Matriz de Integración de TIC en Procesos Educativosjualop100% (1)
- ASISTOLIADocument3 paginiASISTOLIAJackson FloresÎncă nu există evaluări
- Herramientas de AutorDocument11 paginiHerramientas de AutorJoel Jose PinilloÎncă nu există evaluări
- Las Innovaciones Educativas en MéxicoDocument4 paginiLas Innovaciones Educativas en MéxicoNatyta Velez MirandaÎncă nu există evaluări
- Reacciones Adversas Antidiabeticos UltimoDocument66 paginiReacciones Adversas Antidiabeticos Ultimomajo gsÎncă nu există evaluări
- El Valor de La ResponsabilidadDocument1 paginăEl Valor de La ResponsabilidadCésar Emilio Florián PérezÎncă nu există evaluări
- La Nueva Masculinidad y ValoresDocument2 paginiLa Nueva Masculinidad y ValoresIronellys PerezÎncă nu există evaluări
- Tesis TabaquismoDocument91 paginiTesis TabaquismoJaime Estuardo Guzman MoralesÎncă nu există evaluări
- Asa Cardíaca y LateralidadDocument4 paginiAsa Cardíaca y LateralidadHarold AdriánÎncă nu există evaluări
- Mecanismos de Adaptacion Celular.Document14 paginiMecanismos de Adaptacion Celular.pedrolloveraÎncă nu există evaluări
- Política Exterior de Estados Unidos de América Siglo XIXDocument6 paginiPolítica Exterior de Estados Unidos de América Siglo XIXSergio Andrés Ramírez FigueredoÎncă nu există evaluări
- Ensayo Evolucion de La WebDocument4 paginiEnsayo Evolucion de La WebJhoanzend Gerardo Cañamo SÎncă nu există evaluări
- Tema 1 TAD 2Document11 paginiTema 1 TAD 2Fausto PeraltaÎncă nu există evaluări
- Malformaciones Congenitas PDFDocument152 paginiMalformaciones Congenitas PDFJennyLizbethÎncă nu există evaluări
- Morfologia de Las Celulas HematopoyeticasDocument16 paginiMorfologia de Las Celulas HematopoyeticasYanibel FrancoÎncă nu există evaluări
- Iinmunidad Frente A Bacterias IntracelularesDocument51 paginiIinmunidad Frente A Bacterias IntracelularesElsa ParedesÎncă nu există evaluări
- Alcalosis y AcidosisDocument6 paginiAlcalosis y AcidosisAnaParedesÎncă nu există evaluări
- Guia Didactica Dibujo Técnico - Planos TopograficosDocument7 paginiGuia Didactica Dibujo Técnico - Planos TopograficosmariajoseÎncă nu există evaluări
- Seminario de Leucemia CorregidoDocument22 paginiSeminario de Leucemia CorregidoAndreaÎncă nu există evaluări
- Estupor y Estado de ComaDocument26 paginiEstupor y Estado de ComaKary LimaÎncă nu există evaluări
- Definició N de TicDocument36 paginiDefinició N de TicwillnerÎncă nu există evaluări
- Diapositiva Piel y Anexos Cutáneos IIDocument13 paginiDiapositiva Piel y Anexos Cutáneos IIYeison AcendraÎncă nu există evaluări
- Dominio CognoscitivoDocument3 paginiDominio CognoscitivokatharsisdiÎncă nu există evaluări
- Curso de Capacitación DocenteDocument5 paginiCurso de Capacitación DocenteDiana DuránÎncă nu există evaluări
- Formato de Historia Clínica PREGRADODocument6 paginiFormato de Historia Clínica PREGRADOhederÎncă nu există evaluări
- Plexos CoroideosDocument4 paginiPlexos CoroideosJahaira Luna VictoriaÎncă nu există evaluări
- Miembro FantasmaDocument7 paginiMiembro FantasmaJavieraFernandaÎncă nu există evaluări
- TicsDocument13 paginiTicsperlafloresarellanoÎncă nu există evaluări
- La Web Semantica en EducacionDocument12 paginiLa Web Semantica en EducacionLoadUpOnGunsÎncă nu există evaluări
- Rsierra - 1.1 Ensayo, Plataformas VirtualesDocument12 paginiRsierra - 1.1 Ensayo, Plataformas VirtualesRodo SierraÎncă nu există evaluări
- Realidad Aumentada 2.0Document3 paginiRealidad Aumentada 2.0Jairo Nicolas Rivera BayonaÎncă nu există evaluări
- Conclusiones Modelos de Diseño InstruccionalDocument3 paginiConclusiones Modelos de Diseño InstruccionalBrisceyda Arce BojórquezÎncă nu există evaluări
- Tractos EspinotalamicoDocument6 paginiTractos EspinotalamicoGerardo FloresÎncă nu există evaluări
- Practica Sistema Reproductor MasculinoDocument8 paginiPractica Sistema Reproductor MasculinoMirian Antonio rojasÎncă nu există evaluări
- Leucemia TrabajoDocument26 paginiLeucemia TrabajoMichael ArianÎncă nu există evaluări
- Dogma Central de La BiologíaDocument3 paginiDogma Central de La BiologíaKatherine TorresÎncă nu există evaluări
- Absceso CerebralDocument7 paginiAbsceso Cerebralwww.pacourgencias.blogspot.com100% (1)
- Instrumento de Evaluación de Un Afiche SonidoDocument2 paginiInstrumento de Evaluación de Un Afiche SonidoAntonia Rodriguez UrraÎncă nu există evaluări
- Polineuropatias Diagnostico DiferencialDocument102 paginiPolineuropatias Diagnostico DiferencialmarlingaxÎncă nu există evaluări
- Protocolo (Influencia de La Televicion)Document8 paginiProtocolo (Influencia de La Televicion)llopez_329Încă nu există evaluări
- Garrapatas (Ixodidae) II Papel PatógenoDocument7 paginiGarrapatas (Ixodidae) II Papel PatógenoNatha VargasÎncă nu există evaluări
- Herencia MultifactorialDocument23 paginiHerencia MultifactorialDra Daniela TorresÎncă nu există evaluări
- Uso de Las TicsDocument1 paginăUso de Las TicsNatali Sivana Laguna HuancaÎncă nu există evaluări
- Células DentríticasDocument7 paginiCélulas DentríticasNicole Anahi Montes RomeroÎncă nu există evaluări
- Pilares de Una Retroalimentacion EfectivaDocument8 paginiPilares de Una Retroalimentacion EfectivaRebeca RivasÎncă nu există evaluări
- Infertilidad MasculinaDocument14 paginiInfertilidad Masculinaeberth22Încă nu există evaluări
- Hemorragia IntraventricularDocument20 paginiHemorragia IntraventricularMarco Antonio Bautista RamirezÎncă nu există evaluări
- PetequiasDocument2 paginiPetequiasOh UhÎncă nu există evaluări
- GPC CA ProstataDocument11 paginiGPC CA Prostatacerbero36Încă nu există evaluări
- 2011 La Planificacion Didáctica y El Diseño Instruccional en Ambientes de AprendizajeDocument33 pagini2011 La Planificacion Didáctica y El Diseño Instruccional en Ambientes de AprendizajeemelbarackÎncă nu există evaluări
- Articulo Proyecto JENNIFERDocument6 paginiArticulo Proyecto JENNIFERjorge hernandezÎncă nu există evaluări
- Seminario 1. MiocardiopatíasDocument39 paginiSeminario 1. MiocardiopatíasFernando Martín Arce AlvaÎncă nu există evaluări
- Estrabismo 150615033809 Lva1 App6892Document24 paginiEstrabismo 150615033809 Lva1 App6892Oscar Cortez HerreraÎncă nu există evaluări
- TanatologiaDocument17 paginiTanatologiaOscar Cortez HerreraÎncă nu există evaluări
- Diarruda Aguda InfecciosaDocument5 paginiDiarruda Aguda InfecciosaOscar Cortez HerreraÎncă nu există evaluări
- TanatologiaDocument17 paginiTanatologiaOscar Cortez HerreraÎncă nu există evaluări
- Anemia Microcítica Con Caso ClinicoDocument7 paginiAnemia Microcítica Con Caso ClinicoOscar Cortez HerreraÎncă nu există evaluări
- .Control PrenatalDocument83 pagini.Control PrenatalleyrowenaÎncă nu există evaluări
- ZikaDocument24 paginiZikaOscar Cortez HerreraÎncă nu există evaluări
- Guía de DiagnositcoDocument31 paginiGuía de DiagnositcoOscar Cortez HerreraÎncă nu există evaluări
- CefaleasDocument27 paginiCefaleasOscar Cortez HerreraÎncă nu există evaluări
- Medint Gastroenteritis 121111200701 Phpapp02Document29 paginiMedint Gastroenteritis 121111200701 Phpapp02Oscar Cortez HerreraÎncă nu există evaluări
- CefaleasDocument27 paginiCefaleasOscar Cortez HerreraÎncă nu există evaluări
- Apendicitis A Gud ADocument79 paginiApendicitis A Gud AOscar Cortez HerreraÎncă nu există evaluări
- AnemiaferropnicaDocument32 paginiAnemiaferropnicaOscar Cortez HerreraÎncă nu există evaluări
- MACRÓLIDOSDocument7 paginiMACRÓLIDOSinextremus_cayado7754Încă nu există evaluări
- Monografia ParasitoDocument25 paginiMonografia ParasitoOscar Cortez HerreraÎncă nu există evaluări
- Amenaza de Parto PreterminoDocument41 paginiAmenaza de Parto PreterminoOscar Cortez HerreraÎncă nu există evaluări
- Agrotoxicos-Agroquímicos Prohibidos Nacionales e Internacionales-ResumenDocument29 paginiAgrotoxicos-Agroquímicos Prohibidos Nacionales e Internacionales-ResumenRubenAlvarez100% (1)
- AMC - Registro Al XXVI Verano de La Investigación Científica - (Julián Adrián - Mellado)Document11 paginiAMC - Registro Al XXVI Verano de La Investigación Científica - (Julián Adrián - Mellado)julian adrianÎncă nu există evaluări
- Los TrabalenguasDocument10 paginiLos TrabalenguasAngello CalderonÎncă nu există evaluări
- HEMATOLOGIADocument5 paginiHEMATOLOGIACátiaCristinaÎncă nu există evaluări
- Actividades de Repaso Unidad 2 - 1ºESODocument3 paginiActividades de Repaso Unidad 2 - 1ºESOMaría del Carmen Bedón FuentesÎncă nu există evaluări
- Matriz de Riesgo Drogueria Zamar 1Document3 paginiMatriz de Riesgo Drogueria Zamar 1Leidy Martinez100% (2)
- Ubicuidad y Diversidad MicrobianaDocument13 paginiUbicuidad y Diversidad MicrobianaaracelyÎncă nu există evaluări
- Neuro CienciaDocument114 paginiNeuro CienciaJoseAlfaroDíazÎncă nu există evaluări
- Tesis-Enfermería-2019-Condori Jurado y Ramos LopezDocument86 paginiTesis-Enfermería-2019-Condori Jurado y Ramos LopezCarlitos_Valera_romeÎncă nu există evaluări
- Di-Sst-07 Acta CopasstDocument5 paginiDi-Sst-07 Acta CopasstLIZA SSTÎncă nu există evaluări
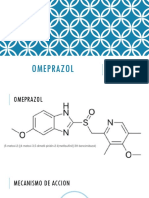
- Omeprazol ExposicionDocument10 paginiOmeprazol Exposicion徳利雅Încă nu există evaluări
- Plan de Negocios Fisiomeds Melendez, Garat, Schulz Rev MZM Corregido 2Document141 paginiPlan de Negocios Fisiomeds Melendez, Garat, Schulz Rev MZM Corregido 2David Schultz CortesÎncă nu există evaluări
- Notas de PresentacionesDocument8 paginiNotas de PresentacionesfabiolaÎncă nu există evaluări
- Memoria de Calculo Unidades EducativasDocument51 paginiMemoria de Calculo Unidades EducativasJimmy VinoÎncă nu există evaluări
- Capitulo 13 Arritmias CardiacasDocument11 paginiCapitulo 13 Arritmias CardiacasTax KaryÎncă nu există evaluări
- Examen MicrobioDocument8 paginiExamen MicrobioJose Ignacio Lopez Garcia100% (1)
- Manejo de Productos Químicos en La ConstrucciónDocument3 paginiManejo de Productos Químicos en La Construccióntavo dominguezÎncă nu există evaluări
- AROMATERAPIA Clase 6 BuenaVidaDocument8 paginiAROMATERAPIA Clase 6 BuenaVidaRenacer Terapias HolisticasÎncă nu există evaluări
- Patologia de Torax y AbdomenDocument9 paginiPatologia de Torax y AbdomenSoledad ColqueÎncă nu există evaluări
- Inventario de Pensamientos Automáticos Ruiz y LujanDocument3 paginiInventario de Pensamientos Automáticos Ruiz y LujanRibertx Del Carpio Hernandez100% (2)
- Actividades para Articular RRDocument14 paginiActividades para Articular RRJAIME DANIEL JARAMILLO JUMBOÎncă nu există evaluări
- 4.sindromes PleuralesDocument23 pagini4.sindromes PleuralesAlvaro Rodrigo Benites OrderiqueÎncă nu există evaluări
- Triptico EpilepsiaDocument2 paginiTriptico EpilepsiaEli CuauhizoÎncă nu există evaluări
- Niveles de Lengua - Rolfi RamirezDocument5 paginiNiveles de Lengua - Rolfi RamirezRosaura Ramirez OrtizÎncă nu există evaluări
- Juan Davila D6Document5 paginiJuan Davila D6pepedlpollo1100% (2)
- Antiinflamatorios EsteroideosDocument38 paginiAntiinflamatorios EsteroideosRodrigo NavaÎncă nu există evaluări
- Análisis de Peligros Higiénicos en Un Proceso de Soldadura.Document13 paginiAnálisis de Peligros Higiénicos en Un Proceso de Soldadura.Margarita Vergara Salcedo100% (2)
- Dermatitis Por Contacto AlergicaDocument17 paginiDermatitis Por Contacto AlergicaJosefina LomeliÎncă nu există evaluări
- Aparato Locomotor Primera - SemanaDocument4 paginiAparato Locomotor Primera - SemanasandraÎncă nu există evaluări
- 7 AdenopatiasDocument4 pagini7 Adenopatiasleonor1862Încă nu există evaluări