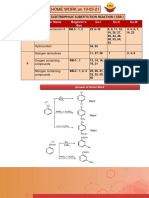
S-ar putea să vă placă și
- Handbook of Coordination Catalysis in Organic ChemistryDe la EverandHandbook of Coordination Catalysis in Organic ChemistryÎncă nu există evaluări
- Experiment 1 SCH2413 PDFDocument4 paginiExperiment 1 SCH2413 PDFSyuhada AhmadÎncă nu există evaluări
- Critical Evaluation of Equilibrium Constants Involving 8-Hydroxyquinoline and Its Metal Chelates: Critical Evaluation of Equilibrium Constants in Solution: Part B: Equilibrium Constants of Liquid-Liquid Distribution SystemsDe la EverandCritical Evaluation of Equilibrium Constants Involving 8-Hydroxyquinoline and Its Metal Chelates: Critical Evaluation of Equilibrium Constants in Solution: Part B: Equilibrium Constants of Liquid-Liquid Distribution SystemsÎncă nu există evaluări
- Laboratory 1 - Alkyl HalidesDocument7 paginiLaboratory 1 - Alkyl Halidessindhsanam100% (1)
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesDe la EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesÎncă nu există evaluări
- Haloalkanes Haloarenes Bounce BackDocument166 paginiHaloalkanes Haloarenes Bounce BackShivadeep Vishwakarma100% (1)
- Laboratory 1 - Alkyl HalidesDocument7 paginiLaboratory 1 - Alkyl HalidesbidinÎncă nu există evaluări
- Chimie GB 2013 FinalDocument11 paginiChimie GB 2013 FinalChu Thi Hien ThuÎncă nu există evaluări
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesDe la EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesÎncă nu există evaluări
- Block 6 Functional Groups 2Document46 paginiBlock 6 Functional Groups 2Cheng FuÎncă nu există evaluări
- Haloalkanes & HaloarenesDocument10 paginiHaloalkanes & Haloarenesakshatshukla2021Încă nu există evaluări
- A2 Chemistry ExamzoneDocument4 paginiA2 Chemistry ExamzoneSan SiddzÎncă nu există evaluări
- Phenol Synthesis via Cumene Hydroperoxide Cleavage (Hock MethodDocument10 paginiPhenol Synthesis via Cumene Hydroperoxide Cleavage (Hock MethodRizkyanto NugrohoÎncă nu există evaluări
- Acfrogbyyb W54zpzfswkn8k3vq0clq6et8mk Ne Px62hvrlk5chrlql9xx83xtq2sr0dqcuhrswcoglr Ueky068cras4ph7jxkmy 143kq0wnhekbynbh 4 Eq1p0kvslajoriecir6ikqqswDocument8 paginiAcfrogbyyb W54zpzfswkn8k3vq0clq6et8mk Ne Px62hvrlk5chrlql9xx83xtq2sr0dqcuhrswcoglr Ueky068cras4ph7jxkmy 143kq0wnhekbynbh 4 Eq1p0kvslajoriecir6ikqqswThanh Hằng NgôÎncă nu există evaluări
- Absorption of Carbon Dioxide in Packed Column: Praveen.S.Nair, P.P.SelviDocument11 paginiAbsorption of Carbon Dioxide in Packed Column: Praveen.S.Nair, P.P.SelviAvuyile KalityiÎncă nu există evaluări
- Halogenalkanes: Unit 2 Chemistry C. Bailey PolackDocument23 paginiHalogenalkanes: Unit 2 Chemistry C. Bailey PolackBritney PattersonÎncă nu există evaluări
- Halogenalkanes: Unit 2 Chemistry C. Bailey PolackDocument23 paginiHalogenalkanes: Unit 2 Chemistry C. Bailey PolackBritney PattersonÎncă nu există evaluări
- Thelmechanisms: of Reductive Carboxylation ReactionsDocument8 paginiThelmechanisms: of Reductive Carboxylation ReactionsRaymond LaBoyÎncă nu există evaluări
- 4.3 Alkyhalide PreparationDocument23 pagini4.3 Alkyhalide PreparationDawit BirhanuÎncă nu există evaluări
- Halogen DerivativesDocument11 paginiHalogen DerivativesJayjayjay 5100% (1)
- Nucleophilic SubstitutionDocument5 paginiNucleophilic SubstitutionNikki Francine BaldeÎncă nu există evaluări
- Chemistry Test - 12th Science-ChemistryDocument7 paginiChemistry Test - 12th Science-ChemistryAishley ChalametÎncă nu există evaluări
- Exp 10 Alkyl HalidesDocument18 paginiExp 10 Alkyl HalidesGeorge PiliposyanÎncă nu există evaluări
- 12 Chemistry Keypoints Revision Questions Chapter 10 PDFDocument13 pagini12 Chemistry Keypoints Revision Questions Chapter 10 PDFSahil Kalra100% (1)
- CHE3044F, 2013: Reactor Design 1: TUTORIAL 8: K 0.1 Mi N K 0.1 Mi NDocument2 paginiCHE3044F, 2013: Reactor Design 1: TUTORIAL 8: K 0.1 Mi N K 0.1 Mi NnmhatityeÎncă nu există evaluări
- HW 2 2007Document4 paginiHW 2 2007Singh AnujÎncă nu există evaluări
- Experiment 12Document17 paginiExperiment 12Yvince LohÎncă nu există evaluări
- Slide 1Document26 paginiSlide 1ShreyaÎncă nu există evaluări
- 5.2.3 Redox and Electrode Potentials QPDocument37 pagini5.2.3 Redox and Electrode Potentials QPUbayd HussainÎncă nu există evaluări
- BCHCT 133Document16 paginiBCHCT 133Md YusufÎncă nu există evaluări
- SN 2Document3 paginiSN 2amel saadÎncă nu există evaluări
- Seminar 28Document31 paginiSeminar 28Sunil PillaiÎncă nu există evaluări
- Preparation of Alkyl Halides, R-X Reaction of Alkanes With CL & BR (F Is Too Reactive, I Is Unreactive)Document20 paginiPreparation of Alkyl Halides, R-X Reaction of Alkanes With CL & BR (F Is Too Reactive, I Is Unreactive)davidtomyÎncă nu există evaluări
- Wolaita Sodo University: General Chemistry (Chem.1012) Chapter FourDocument56 paginiWolaita Sodo University: General Chemistry (Chem.1012) Chapter FourAbdulmajid AbdellaÎncă nu există evaluări
- CHEMISTRY Questions - 2019-20 - SET1Document8 paginiCHEMISTRY Questions - 2019-20 - SET1-Uddipan BagchiÎncă nu există evaluări
- Le Chatelier's Principle - Chromate-Dichromate - C12!4!07Document5 paginiLe Chatelier's Principle - Chromate-Dichromate - C12!4!07John Michael Maulas Vargas100% (1)
- Major Organic Reactions ChapterDocument63 paginiMajor Organic Reactions ChapterdagmawiÎncă nu există evaluări
- Section A: Mcqs Halogen DerivativesDocument11 paginiSection A: Mcqs Halogen DerivativesBint A. Qadir100% (1)
- Distinction Test (Organic Chemistry)Document14 paginiDistinction Test (Organic Chemistry)Ashish Kumar100% (3)
- Haloalkanes and HaloarenesDocument14 paginiHaloalkanes and HaloarenesHarshitha GowdaÎncă nu există evaluări
- Nucleophilic Substitution Questions - PKBDocument12 paginiNucleophilic Substitution Questions - PKBPawan BabelÎncă nu există evaluări
- Experimental Procedures General Chemistry I KI-1101Document21 paginiExperimental Procedures General Chemistry I KI-1101Danni SulaimanÎncă nu există evaluări
- Optimization in The Absorption and DesorptionDocument20 paginiOptimization in The Absorption and DesorptionShamsMohdÎncă nu există evaluări
- Mass Balances With Reactions 4.1-4.3 ExercisesDocument8 paginiMass Balances With Reactions 4.1-4.3 ExercisesJackson MakgolengÎncă nu există evaluări
- HaloalkanesDocument13 paginiHaloalkanesChingYan TanÎncă nu există evaluări
- CHAPTER 6 Alkyl Halides and Aryl HalidesDocument150 paginiCHAPTER 6 Alkyl Halides and Aryl HalidesexpertwritersÎncă nu există evaluări
- Reduction Reactions Mechanisms and SelectivityDocument34 paginiReduction Reactions Mechanisms and SelectivityFaTin AziEyatiÎncă nu există evaluări
- II PUC Chemistry - SN1 and SN2 MechanismsDocument4 paginiII PUC Chemistry - SN1 and SN2 MechanismsShamanth MÎncă nu există evaluări
- Chemical Reaction Engineering TutorialDocument13 paginiChemical Reaction Engineering TutorialNg Joshua0% (1)
- 1Document6 pagini170123Încă nu există evaluări
- Inorganic Lab Exp 2Document6 paginiInorganic Lab Exp 2Jekyll Rev67% (3)
- Chm096 Chapter 2 Chemical Kinetics Nov 2013 - Mac 2014Document167 paginiChm096 Chapter 2 Chemical Kinetics Nov 2013 - Mac 2014Irsyad KamilÎncă nu există evaluări
- Cyanex 272Document37 paginiCyanex 272partyweirdo100% (2)
- Synergistic Catalysis by Lewis Acid and Base Sites On Zro For Meerwein Ponndorf Verley ReductionDocument7 paginiSynergistic Catalysis by Lewis Acid and Base Sites On Zro For Meerwein Ponndorf Verley ReductionRiza SaidÎncă nu există evaluări
- Experiment 4Document7 paginiExperiment 4Pratik PatelÎncă nu există evaluări
- Synthesis of Quinolizinium SaltsDocument3 paginiSynthesis of Quinolizinium Saltsspkgchem74670% (1)
- Ways To Overcome Unethical Chemical Waste DisposalDocument4 paginiWays To Overcome Unethical Chemical Waste DisposalbidinÎncă nu există evaluări
- CHEM-2202 (3) - 072 Sofia Soriano Student# 302-588-9Document6 paginiCHEM-2202 (3) - 072 Sofia Soriano Student# 302-588-9bidinÎncă nu există evaluări
- Discurso de Werner, Ganador de Premio Nobel 1913Document14 paginiDiscurso de Werner, Ganador de Premio Nobel 1913Angélica SixtosÎncă nu există evaluări
- 1) The Structure and Foundation of Ethics in Islam1Document48 pagini1) The Structure and Foundation of Ethics in Islam1bidinÎncă nu există evaluări
- Lecture Water Activity-Part 1Document24 paginiLecture Water Activity-Part 1bidinÎncă nu există evaluări
- CHEM-2202 (3) - 072 Sofia Soriano Student# 302-588-9Document6 paginiCHEM-2202 (3) - 072 Sofia Soriano Student# 302-588-9bidinÎncă nu există evaluări
- W 14 PhenolsDocument53 paginiW 14 Phenolsbidin100% (1)
- Atomic Structure and Fundamental ParticlesDocument32 paginiAtomic Structure and Fundamental ParticleshumayunbashaÎncă nu există evaluări
- 06.-Medidor Cloro CL4000enDocument18 pagini06.-Medidor Cloro CL4000enLeonel RubioÎncă nu există evaluări
- Inline Desilter ManualDocument18 paginiInline Desilter ManualdesaviniciusÎncă nu există evaluări
- CO2 Depressurisation OLGADocument8 paginiCO2 Depressurisation OLGAMaheshÎncă nu există evaluări
- Make a castable lab test mixDocument16 paginiMake a castable lab test mixthaituan237Încă nu există evaluări
- Teknik Menjawab KIMIA 2011Document73 paginiTeknik Menjawab KIMIA 2011Nur HakimÎncă nu există evaluări
- Magic Equation r1 (All Codes)Document16 paginiMagic Equation r1 (All Codes)Prashant SunagarÎncă nu există evaluări
- MIT2 080JF13 Lecture2 PDFDocument26 paginiMIT2 080JF13 Lecture2 PDFAbhilashJanaÎncă nu există evaluări
- Egee 101 Reflective Essay 1Document3 paginiEgee 101 Reflective Essay 1api-142590237Încă nu există evaluări
- Chapter 1 - Introduction To Differential EquationsDocument16 paginiChapter 1 - Introduction To Differential EquationsnawidwardakÎncă nu există evaluări
- Extrusion Press PDFDocument2 paginiExtrusion Press PDFRobert25% (4)
- The Weighted Histogram Analysis Method (WHAM) : Michael AndrecDocument14 paginiThe Weighted Histogram Analysis Method (WHAM) : Michael AndrecWilliam AgudeloÎncă nu există evaluări
- Review of Literature On Probability of Detection For Liquid Penetrant Nondestructive TestingDocument51 paginiReview of Literature On Probability of Detection For Liquid Penetrant Nondestructive TestingVicky GautamÎncă nu există evaluări
- E-DWT-H Electronic Deadweight TesterDocument2 paginiE-DWT-H Electronic Deadweight TesterMorosanu Andreea-DianaÎncă nu există evaluări
- Modeling of Synchronous Generators in Power System Studies: October 2016Document12 paginiModeling of Synchronous Generators in Power System Studies: October 2016aswardiÎncă nu există evaluări
- Vibration assignment on 3DOF cantilever beam systemDocument5 paginiVibration assignment on 3DOF cantilever beam systemIzzah 'AtirahÎncă nu există evaluări
- Centrifugal CastingDocument266 paginiCentrifugal Castinguzairmetallurgist100% (2)
- Experimental Investigation of Process Parameters On Inconel 925 For EDM Process by Using Taguchi MethodDocument6 paginiExperimental Investigation of Process Parameters On Inconel 925 For EDM Process by Using Taguchi MethodVishal Kumar JaiswalÎncă nu există evaluări
- How Does DCPIP WorkDocument3 paginiHow Does DCPIP WorkIsaac LeeÎncă nu există evaluări
- Geometric TolerancesDocument6 paginiGeometric Tolerancesvaibhavgitevaibhav_9Încă nu există evaluări
- Design Project: SEV200 - Geotechnical Investigation and Design Last Update: 09/05/2020Document12 paginiDesign Project: SEV200 - Geotechnical Investigation and Design Last Update: 09/05/2020abdulqadirghoriÎncă nu există evaluări
- Structural Health Monitoring: Abin Paul Roll No:4 S7, CE-ADocument34 paginiStructural Health Monitoring: Abin Paul Roll No:4 S7, CE-AAnjana kpÎncă nu există evaluări
- Control System (136-248) PDFDocument113 paginiControl System (136-248) PDFmuruganÎncă nu există evaluări
- 1 4 Bookmark A Crash Course in Forces MotionDocument2 pagini1 4 Bookmark A Crash Course in Forces Motionapi-115513756Încă nu există evaluări
- Amateur's Telescope Was First Published in 1920. However, Unlike Ellison's TimeDocument4 paginiAmateur's Telescope Was First Published in 1920. However, Unlike Ellison's Timemohamadazaresh0% (1)
- Helm (2008) : Section 32.4: Parabolic PdesDocument24 paginiHelm (2008) : Section 32.4: Parabolic Pdestarek mahmoudÎncă nu există evaluări
- Filtration of WaterDocument32 paginiFiltration of WaterYusuf Rahmat SidikÎncă nu există evaluări
- Cara SamplingDocument8 paginiCara SamplingAngga Dwi PutrantoÎncă nu există evaluări
- Electrostatic Discharge Ignition of Energetic MaterialsDocument9 paginiElectrostatic Discharge Ignition of Energetic Materialspamos1111Încă nu există evaluări
- Bhavans Public School, Doha - Qatar: Model Question Paper 2016-17 MathematicsDocument4 paginiBhavans Public School, Doha - Qatar: Model Question Paper 2016-17 MathematicsSanthosh KrishnanÎncă nu există evaluări
- The Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaDe la EverandThe Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaÎncă nu există evaluări
- Sully: The Untold Story Behind the Miracle on the HudsonDe la EverandSully: The Untold Story Behind the Miracle on the HudsonEvaluare: 4 din 5 stele4/5 (101)
- Highest Duty: My Search for What Really MattersDe la EverandHighest Duty: My Search for What Really MattersÎncă nu există evaluări
- Transformed: Moving to the Product Operating ModelDe la EverandTransformed: Moving to the Product Operating ModelEvaluare: 4 din 5 stele4/5 (1)
- Dirt to Soil: One Family’s Journey into Regenerative AgricultureDe la EverandDirt to Soil: One Family’s Journey into Regenerative AgricultureEvaluare: 5 din 5 stele5/5 (124)
- Pale Blue Dot: A Vision of the Human Future in SpaceDe la EverandPale Blue Dot: A Vision of the Human Future in SpaceEvaluare: 4.5 din 5 stele4.5/5 (586)
- The Fabric of Civilization: How Textiles Made the WorldDe la EverandThe Fabric of Civilization: How Textiles Made the WorldEvaluare: 4.5 din 5 stele4.5/5 (57)
- A Place of My Own: The Architecture of DaydreamsDe la EverandA Place of My Own: The Architecture of DaydreamsEvaluare: 4 din 5 stele4/5 (241)
- The Technology Trap: Capital, Labor, and Power in the Age of AutomationDe la EverandThe Technology Trap: Capital, Labor, and Power in the Age of AutomationEvaluare: 4.5 din 5 stele4.5/5 (46)
- 35 Miles From Shore: The Ditching and Rescue of ALM Flight 980De la Everand35 Miles From Shore: The Ditching and Rescue of ALM Flight 980Evaluare: 4 din 5 stele4/5 (21)
- Data-ism: The Revolution Transforming Decision Making, Consumer Behavior, and Almost Everything ElseDe la EverandData-ism: The Revolution Transforming Decision Making, Consumer Behavior, and Almost Everything ElseEvaluare: 3.5 din 5 stele3.5/5 (12)
- Faster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestDe la EverandFaster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestEvaluare: 4 din 5 stele4/5 (28)
- Recording Unhinged: Creative and Unconventional Music Recording TechniquesDe la EverandRecording Unhinged: Creative and Unconventional Music Recording TechniquesÎncă nu există evaluări
- Packing for Mars: The Curious Science of Life in the VoidDe la EverandPacking for Mars: The Curious Science of Life in the VoidEvaluare: 4 din 5 stele4/5 (1395)
- The Future of Geography: How the Competition in Space Will Change Our WorldDe la EverandThe Future of Geography: How the Competition in Space Will Change Our WorldEvaluare: 4.5 din 5 stele4.5/5 (4)
- Across the Airless Wilds: The Lunar Rover and the Triumph of the Final Moon LandingsDe la EverandAcross the Airless Wilds: The Lunar Rover and the Triumph of the Final Moon LandingsÎncă nu există evaluări
- The Mushroom at the End of the World: On the Possibility of Life in Capitalist RuinsDe la EverandThe Mushroom at the End of the World: On the Possibility of Life in Capitalist RuinsEvaluare: 4 din 5 stele4/5 (139)
- The Big, Bad Book of Botany: The World's Most Fascinating FloraDe la EverandThe Big, Bad Book of Botany: The World's Most Fascinating FloraEvaluare: 3 din 5 stele3/5 (10)
- A Garden of Marvels: How We Discovered that Flowers Have Sex, Leaves Eat Air, and Other Secrets of PlantsDe la EverandA Garden of Marvels: How We Discovered that Flowers Have Sex, Leaves Eat Air, and Other Secrets of PlantsÎncă nu există evaluări
- ChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindDe la EverandChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindÎncă nu există evaluări
- The Things We Make: The Unknown History of Invention from Cathedrals to Soda CansDe la EverandThe Things We Make: The Unknown History of Invention from Cathedrals to Soda CansÎncă nu există evaluări
- Inventor of the Future: The Visionary Life of Buckminster FullerDe la EverandInventor of the Future: The Visionary Life of Buckminster FullerEvaluare: 4 din 5 stele4/5 (10)
- Fallout: The Hiroshima Cover-up and the Reporter Who Revealed It to the WorldDe la EverandFallout: The Hiroshima Cover-up and the Reporter Who Revealed It to the WorldEvaluare: 4.5 din 5 stele4.5/5 (82)
- Reality+: Virtual Worlds and the Problems of PhilosophyDe la EverandReality+: Virtual Worlds and the Problems of PhilosophyEvaluare: 4 din 5 stele4/5 (24)
- Einstein's Fridge: How the Difference Between Hot and Cold Explains the UniverseDe la EverandEinstein's Fridge: How the Difference Between Hot and Cold Explains the UniverseEvaluare: 4.5 din 5 stele4.5/5 (50)
- The End of Craving: Recovering the Lost Wisdom of Eating WellDe la EverandThe End of Craving: Recovering the Lost Wisdom of Eating WellEvaluare: 4.5 din 5 stele4.5/5 (80)