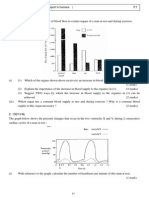
S-ar putea să vă placă și
- Handbook of Critical Care Nephrology 2021Document851 paginiHandbook of Critical Care Nephrology 2021Roberto Barbery100% (3)
- Your BodyDocument19 paginiYour Bodybbe43196% (25)
- How To Improve Circulation With HomeopathyDocument3 paginiHow To Improve Circulation With HomeopathyamazingdivinegraceÎncă nu există evaluări
- Congenital Lung MalformationsDocument115 paginiCongenital Lung MalformationsAhmad Abu KushÎncă nu există evaluări
- Summary (Trematodes)Document4 paginiSummary (Trematodes)Krizia Del Rosario50% (2)
- Procedure-Central Venous Access Catheter InsertionDocument18 paginiProcedure-Central Venous Access Catheter Insertionmohamad dildarÎncă nu există evaluări
- Aplastic Anemia - An Overview: DR Aniruddh Shrivastava Guided By: DR S.H. Talib SIRDocument42 paginiAplastic Anemia - An Overview: DR Aniruddh Shrivastava Guided By: DR S.H. Talib SIRdoctoranswerit_84161Încă nu există evaluări
- Approach To HematuriaDocument45 paginiApproach To HematuriaArun GeorgeÎncă nu există evaluări
- Renal Path Q'sDocument20 paginiRenal Path Q'skank_sÎncă nu există evaluări
- Mjhid 7 1 E2015036Document3 paginiMjhid 7 1 E2015036Admin neuro-usu.idÎncă nu există evaluări
- Spontaneous Rupture of The Kidney Due To Acute Lymphoblastic Leukemia: The First Episode and A Glance On The LiteratureDocument4 paginiSpontaneous Rupture of The Kidney Due To Acute Lymphoblastic Leukemia: The First Episode and A Glance On The LiteraturePeertechz Publications Inc.Încă nu există evaluări
- PM2020 I+NephrologyDocument201 paginiPM2020 I+Nephrologychongyu888xiongÎncă nu există evaluări
- Original Article: Megaloblastic Anemia With Hypotension and Transient Delirium As The Primary Symptoms: Report of A CaseDocument5 paginiOriginal Article: Megaloblastic Anemia With Hypotension and Transient Delirium As The Primary Symptoms: Report of A CaseNindy PutriÎncă nu există evaluări
- BCR 2012 007205Document4 paginiBCR 2012 007205shofidhiaaaÎncă nu există evaluări
- Ischemic Liver InjuryDocument2 paginiIschemic Liver InjuryDanuÎncă nu există evaluări
- 10clinical VignettespdfDocument71 pagini10clinical VignettespdfKerin Ardy100% (1)
- Gastric Antral Vascular Ectasia - Case Report and Literature ReviewDocument5 paginiGastric Antral Vascular Ectasia - Case Report and Literature ReviewHayin NailaÎncă nu există evaluări
- Gi 4Document3 paginiGi 4Syifa' FauziyahÎncă nu există evaluări
- Step by Step: Clinical Problem-SolvingDocument5 paginiStep by Step: Clinical Problem-SolvingMirin ApuniusÎncă nu există evaluări
- Systemic AmiloidosisDocument4 paginiSystemic AmiloidosisioannesturrisoricisÎncă nu există evaluări
- 2014 Kabir Et Al. - Chronic Eosinophilic Leukaemia Presenting With A CDocument4 pagini2014 Kabir Et Al. - Chronic Eosinophilic Leukaemia Presenting With A CPratyay HasanÎncă nu există evaluări
- SESSION 3 - PATHOPHYSIOLOGY With AnswersDocument9 paginiSESSION 3 - PATHOPHYSIOLOGY With AnswersSpeed SamÎncă nu există evaluări
- Barsoum 2004Document13 paginiBarsoum 2004Bruno MoraesÎncă nu există evaluări
- Utah KidneyDocument16 paginiUtah KidneyChristineGonzalesÎncă nu există evaluări
- A Rare Cause of AA Amyloidosis and End-Stage Kidney Failure: QuestionsDocument3 paginiA Rare Cause of AA Amyloidosis and End-Stage Kidney Failure: QuestionsSezen YılmazÎncă nu există evaluări
- End-Stage Renal Disease With Erythrocytosis: A Case Report On A Rare Association of Crescentic Glomerulonephritis and JAK2-Positive Polycythemia VeraDocument6 paginiEnd-Stage Renal Disease With Erythrocytosis: A Case Report On A Rare Association of Crescentic Glomerulonephritis and JAK2-Positive Polycythemia VeraPolito SoytiÎncă nu există evaluări
- Clinical Presentation: by Dr. Raffiq AbbasDocument36 paginiClinical Presentation: by Dr. Raffiq AbbasKarthick UnleashÎncă nu există evaluări
- Blood Vessels Blood VesselsDocument8 paginiBlood Vessels Blood Vesselsnadasyiriin shaqinazÎncă nu există evaluări
- Pi Is 0272638698001097Document6 paginiPi Is 0272638698001097Mita AdrianiÎncă nu există evaluări
- Nejmcps 1808494Document6 paginiNejmcps 1808494dias100% (1)
- A Rare Cause of Reversible Cardiomyopathy PKPDocument27 paginiA Rare Cause of Reversible Cardiomyopathy PKPlichumo murryÎncă nu există evaluări
- Trousseau's Syndrome in CholangiocarcinomaDocument7 paginiTrousseau's Syndrome in CholangiocarcinomaAnna MariaÎncă nu există evaluări
- Letters To The Editor: Membranous Glomerulonephritis in A Patient With SyphilisDocument2 paginiLetters To The Editor: Membranous Glomerulonephritis in A Patient With SyphilisBetty Romero BarriosÎncă nu există evaluări
- In-Depth Review: Nonocclusive Mesenteric Ischemia: A Lethal Complication in Peritoneal Dialysis PatientsDocument6 paginiIn-Depth Review: Nonocclusive Mesenteric Ischemia: A Lethal Complication in Peritoneal Dialysis PatientsFernandoÎncă nu există evaluări
- Hematom 2Document2 paginiHematom 2omidazadmehr1375Încă nu există evaluări
- Loki CH 1972Document4 paginiLoki CH 1972karemiaÎncă nu există evaluări
- AKI CRDocument3 paginiAKI CRAmeldaÎncă nu există evaluări
- Central Pontine Myelinolysis Without Hyponatraemia: AG Droogan, M Mirakhur, I V Allen, J KirkDocument4 paginiCentral Pontine Myelinolysis Without Hyponatraemia: AG Droogan, M Mirakhur, I V Allen, J KirkANOOPVAÎncă nu există evaluări
- Medical MasterClass MRCP Part 2 Nephrology 2010 ElzohrykDocument82 paginiMedical MasterClass MRCP Part 2 Nephrology 2010 ElzohrykSagvan HajaniÎncă nu există evaluări
- Sme PropofolDocument2 paginiSme PropofolAntonella Jazmín AssadÎncă nu există evaluări
- 01 Renal System NotesDocument22 pagini01 Renal System NotesLexanCantorFermoÎncă nu există evaluări
- PDF TJP 1944Document5 paginiPDF TJP 1944Erlangga D SaputroÎncă nu există evaluări
- Portal Hypertension: Pathophysiology, Diagnosis and ManagementDocument11 paginiPortal Hypertension: Pathophysiology, Diagnosis and ManagementXol-TikaÎncă nu există evaluări
- Caso Clinico Clnica Mayo 3Document6 paginiCaso Clinico Clnica Mayo 3Francisco HernandezÎncă nu există evaluări
- Acute Kidney Injury HELLP SyndromeDocument4 paginiAcute Kidney Injury HELLP SyndromeEllya Latifah IlyasÎncă nu există evaluări
- Universidad Privada San Juan Bautista: Escuela de Medicina HumanaDocument6 paginiUniversidad Privada San Juan Bautista: Escuela de Medicina HumanaKarolLeylaÎncă nu există evaluări
- Colecistitis Aguda Alitiasica: Casos ClinicosDocument3 paginiColecistitis Aguda Alitiasica: Casos Clinicosnahm17Încă nu există evaluări
- Matson 1961Document4 paginiMatson 1961NyimasÎncă nu există evaluări
- ChangDocument3 paginiChangAmirullah AbdiÎncă nu există evaluări
- Priapism - A Rare Presentation in Chronic Myeloid Leukemia: Case Report and Review of The LiteratureDocument5 paginiPriapism - A Rare Presentation in Chronic Myeloid Leukemia: Case Report and Review of The LiteratureSalwiyadiÎncă nu există evaluări
- 2805-01. Manuscript (Article Text) - 6211-1-10-20161202Document3 pagini2805-01. Manuscript (Article Text) - 6211-1-10-20161202Karina Aguirre AlvarezÎncă nu există evaluări
- b3 Journal A Rare CaseDocument2 paginib3 Journal A Rare CaseTessa AcostaÎncă nu există evaluări
- A Treacherous Course: Clinical Problem-SolvingDocument6 paginiA Treacherous Course: Clinical Problem-SolvingMorocco CandycatyÎncă nu există evaluări
- Colitis Isquemica PDFDocument7 paginiColitis Isquemica PDFWaldo ReyesÎncă nu există evaluări
- Two Cases of Variceal Haemorrhage During Living DoDocument3 paginiTwo Cases of Variceal Haemorrhage During Living DoRENAULTÎncă nu există evaluări
- 296 FullDocument4 pagini296 FullDora MukupaÎncă nu există evaluări
- نسخة من HemoglobipathiesDocument63 paginiنسخة من Hemoglobipathiesnour khaleel Mohammad BojaÎncă nu există evaluări
- Caso Clinico Clinica Mayo 2Document5 paginiCaso Clinico Clinica Mayo 2Francisco HernandezÎncă nu există evaluări
- Nephrotic Syndrome in Adults: Acute Medicine 2018 17 (1) : 36-43 36Document8 paginiNephrotic Syndrome in Adults: Acute Medicine 2018 17 (1) : 36-43 36Deddy TriwijayaÎncă nu există evaluări
- Dyspnea With An Abdominal Bruit: Hereditary Hemorrhagic TelangiectasiaDocument2 paginiDyspnea With An Abdominal Bruit: Hereditary Hemorrhagic TelangiectasiaMahmoud AbouelsoudÎncă nu există evaluări
- Hepatocellular Carcinoma Causing Severe.1057Document1 paginăHepatocellular Carcinoma Causing Severe.1057Vardan KocharÎncă nu există evaluări
- Recurrent Unilateral Pleural Effusion From Constrictive Pericarditis of Unknown Etiology Requiring PericardiectomyDocument3 paginiRecurrent Unilateral Pleural Effusion From Constrictive Pericarditis of Unknown Etiology Requiring PericardiectomyJaya Semara PutraÎncă nu există evaluări
- Review: Liver Dysfunction in Critical IllnessDocument17 paginiReview: Liver Dysfunction in Critical IllnessRoger Ludeña SalazarÎncă nu există evaluări
- Infective EndocarditisDocument21 paginiInfective EndocarditisMohamad HamzaÎncă nu există evaluări
- Pi Is 0085253815524253Document5 paginiPi Is 0085253815524253Karina DewataÎncă nu există evaluări
- Left Vs Right: Heart FailureDocument3 paginiLeft Vs Right: Heart FailureRosalinda PerigoÎncă nu există evaluări
- Avian Circulatory SystemDocument27 paginiAvian Circulatory SystemCarlBuscato100% (4)
- Railway Recruitment Board ExaminationDocument51 paginiRailway Recruitment Board ExaminationobroirohitÎncă nu există evaluări
- FlowtronDocument4 paginiFlowtronHari Prasath T RÎncă nu există evaluări
- Schultz 2002Document6 paginiSchultz 2002Rosario SilvaÎncă nu există evaluări
- B7.2 Transport in MammalsDocument13 paginiB7.2 Transport in Mammalsulavan24Încă nu există evaluări
- Final Coaching CardioDocument70 paginiFinal Coaching CardiomarykristiroseÎncă nu există evaluări
- Class 10 TransportationDocument5 paginiClass 10 TransportationKanishk KumarÎncă nu există evaluări
- Guyton at The BedsideDocument7 paginiGuyton at The BedsideGustavo ParedesÎncă nu există evaluări
- Hkcee Biology - 4.6 Transport in Humans - P.1Document13 paginiHkcee Biology - 4.6 Transport in Humans - P.1irisyyy27Încă nu există evaluări
- AD8232 ECG MonitoringDocument7 paginiAD8232 ECG Monitoringsupport ubicuoÎncă nu există evaluări
- A Study To Evaluate Effectiveness of Cold Application and Magnesium Sulphate Application On Superficial Thrombophlebitis Among Patients Receiving Intravenous Therapy in Selected Hospitals Amritsar.Document25 paginiA Study To Evaluate Effectiveness of Cold Application and Magnesium Sulphate Application On Superficial Thrombophlebitis Among Patients Receiving Intravenous Therapy in Selected Hospitals Amritsar.Navjot Brar71% (14)
- Biology Form 2 NotesDocument80 paginiBiology Form 2 NotesLutern RayÎncă nu există evaluări
- Blood Circulation and AnswersDocument2 paginiBlood Circulation and AnswersAyush SinghÎncă nu există evaluări
- First Periodic Test Science ViDocument2 paginiFirst Periodic Test Science ViNick Bantolo67% (9)
- Vicorder List of Tests and Parameters PDFDocument4 paginiVicorder List of Tests and Parameters PDFHatem FaroukÎncă nu există evaluări
- Management of Siraj Granthi (Varicose Vein) Through AyurvedaDocument12 paginiManagement of Siraj Granthi (Varicose Vein) Through AyurvedaPuja ZarkarÎncă nu există evaluări
- Cardiovascular Physiology NodrmDocument528 paginiCardiovascular Physiology NodrmEmmanuel PrahÎncă nu există evaluări
- Lecture 6 Cardiovascular: Vascular System - The HemodynamicsDocument61 paginiLecture 6 Cardiovascular: Vascular System - The HemodynamicsAndreea ŞtefănescuÎncă nu există evaluări
- Total Parenteral Nutrition Peritoneal Dialysis PD Central Venous Pressure CVPDocument3 paginiTotal Parenteral Nutrition Peritoneal Dialysis PD Central Venous Pressure CVPathena19Încă nu există evaluări
- Cardiovascular NotesDocument256 paginiCardiovascular NotesVipul RajoraÎncă nu există evaluări
- Portal Hypertension: Dr. Ravi Gadani MS, FmasDocument57 paginiPortal Hypertension: Dr. Ravi Gadani MS, FmasRaviÎncă nu există evaluări
- Study Material Class XDocument14 paginiStudy Material Class XsaleemÎncă nu există evaluări
- Blood VesselsDocument16 paginiBlood VesselsAlen OsmanovicÎncă nu există evaluări