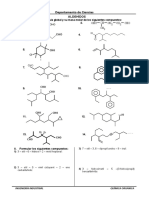
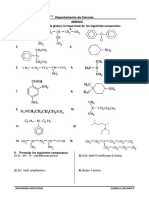
S-ar putea să vă placă și
- Panoptico Rapido Concentrado 1.10Document2 paginiPanoptico Rapido Concentrado 1.10Alejandra100% (1)
- Poe-4 Hemoglobina ClorimetricaDocument8 paginiPoe-4 Hemoglobina ClorimetricaBrido ClarosÎncă nu există evaluări
- Informe de Acidez BromatologiaDocument9 paginiInforme de Acidez BromatologiaJohnAndresPinargoteÎncă nu există evaluări
- Clase 4 Funcion RenalDocument49 paginiClase 4 Funcion RenalJaz Ntvg100% (1)
- Uremia ProspectoDocument3 paginiUremia ProspectoYuMi BautistaÎncă nu există evaluări
- Entamoeba HistolyticaDocument9 paginiEntamoeba HistolyticaOrlando CruzÎncă nu există evaluări
- MacrodantinaDocument8 paginiMacrodantinaangelicaÎncă nu există evaluări
- EgoDocument17 paginiEgoCynthiaÎncă nu există evaluări
- Informe de Liquido AsciticoDocument6 paginiInforme de Liquido AsciticoRosa Merino FernandezÎncă nu există evaluări
- 4 A. - Formas Farmaceuticas Protegidas Por Envoltura 2011-1Document22 pagini4 A. - Formas Farmaceuticas Protegidas Por Envoltura 2011-1Charly Bolivar Benites100% (2)
- Laboratorio OrinaDocument27 paginiLaboratorio OrinaAngie Villar RuizÎncă nu există evaluări
- Anemia Por AlcoholDocument11 paginiAnemia Por AlcoholJazitaRomero100% (1)
- Pract UreaDocument2 paginiPract UreaLesli Lisbeth Molina SotoÎncă nu există evaluări
- Práctica 1Document7 paginiPráctica 1Alejandra Calderón JiménezÎncă nu există evaluări
- Práctica N - 3 CreatininaDocument8 paginiPráctica N - 3 CreatininaJorge CruzÎncă nu există evaluări
- MICODocument52 paginiMICOBriss Carreón CarreónÎncă nu există evaluări
- 4 Orina Clase 9 - Clase 10-Clase 11 Clase 17Document65 pagini4 Orina Clase 9 - Clase 10-Clase 11 Clase 17Marcos BerlingieriÎncă nu există evaluări
- Química Del Proceso de FijaciónDocument52 paginiQuímica Del Proceso de FijaciónAdolfo Antonio Ríos AlcortaÎncă nu există evaluări
- Determinación de Nitrógeno Ureico: Flor Marina Trujillo Girardot Laboratorio ClinicoDocument7 paginiDeterminación de Nitrógeno Ureico: Flor Marina Trujillo Girardot Laboratorio ClinicoMARIA ALEJANDRA OTALORA SANABRIAÎncă nu există evaluări
- 6374 Chagatest Elisa SPDocument3 pagini6374 Chagatest Elisa SPMaría José QuirozÎncă nu există evaluări
- Reporte de Laboratorio CoprocultivoDocument10 paginiReporte de Laboratorio CoprocultivoEMMANUEL MANZO ROSASÎncă nu există evaluări
- Informe 2do Control IbuprofenoDocument11 paginiInforme 2do Control IbuprofenoKarla Quiñones RufinoÎncă nu există evaluări
- GalactosuriaDocument21 paginiGalactosuriaErcelyn Menjivar50% (2)
- Creatinina Enzimatica Aa Liquida SPDocument3 paginiCreatinina Enzimatica Aa Liquida SPKevin ArechigaÎncă nu există evaluări
- QuimicaDocument7 paginiQuimicaYilber RiañoÎncă nu există evaluări
- Hemoclasificación NCDocument8 paginiHemoclasificación NCsergio quinteroÎncă nu există evaluări
- Aseguramineto de La Calidad en Hematologia y HemostasiaDocument27 paginiAseguramineto de La Calidad en Hematologia y HemostasiaBernabéMaqueraQuispe0% (1)
- Caso Clinico GeriatricoDocument4 paginiCaso Clinico GeriatricoPablo Cesar Soto100% (1)
- Amilasa CNPG Liquiform 142 EspDocument6 paginiAmilasa CNPG Liquiform 142 Espelsa ramirezÎncă nu există evaluări
- AQC II Informe # Ácido ÚricoDocument4 paginiAQC II Informe # Ácido Úricoroberth panchanaÎncă nu există evaluări
- Head SpaceDocument52 paginiHead Spaceyesicaberth100% (1)
- Clase 6. Megacariopoyesis y Fisiología PlaquetariaDocument43 paginiClase 6. Megacariopoyesis y Fisiología PlaquetariaEstefany Nicol Saldaña RimarachinÎncă nu există evaluări
- 1 WidalDocument2 pagini1 WidalHans YoveraÎncă nu există evaluări
- Clase N 9 Estudio de Liquidos BiologicosDocument11 paginiClase N 9 Estudio de Liquidos BiologicosMedalyt Huashuayo CusiÎncă nu există evaluări
- TraducciónDocument8 paginiTraducciónMaria FernandaÎncă nu există evaluări
- Tema 02 Produccion y Destruccion EritrocitariaDocument17 paginiTema 02 Produccion y Destruccion EritrocitariaErick Montecinos AlvaradoÎncă nu există evaluări
- Caseina PaperDocument6 paginiCaseina PaperRobin Guevara100% (1)
- Determinacion Cuantitativa Del Acido UricoDocument14 paginiDeterminacion Cuantitativa Del Acido UricoPedro SuarezÎncă nu există evaluări
- Guia Paso Químico de OrinaDocument6 paginiGuia Paso Químico de OrinaElizabeth100% (1)
- T. Sup y OxidacionDocument13 paginiT. Sup y OxidacionROXANA QUISPE MAMANIÎncă nu există evaluări
- Alfa Oxidacion y CetogeneDocument19 paginiAlfa Oxidacion y Cetogenenico0% (1)
- Proteina Total. InsertoDocument2 paginiProteina Total. InsertoFrancisco Baca DejoÎncă nu există evaluări
- Guia de Practica Urinanalisis8Document3 paginiGuia de Practica Urinanalisis8Jhon Andy RamosÎncă nu există evaluări
- Manual InmunohematologíaDocument22 paginiManual InmunohematologíaJuan David GomezÎncă nu există evaluări
- Agar CledDocument1 paginăAgar CledLuis CyferÎncă nu există evaluări
- Farmacologia de Las Vias BiliaresDocument8 paginiFarmacologia de Las Vias BiliaresSelene Calani HerreraÎncă nu există evaluări
- CARAMELIZACIONDocument1 paginăCARAMELIZACIONMarco JuniorÎncă nu există evaluări
- Actualización de Uroanalisis.Document9 paginiActualización de Uroanalisis.Franco Chavez MartinezÎncă nu există evaluări
- CA Arsenazo Liquiform 95 EspDocument6 paginiCA Arsenazo Liquiform 95 Espelsa ramirezÎncă nu există evaluări
- Valoración Química de Drogas VegetalesDocument6 paginiValoración Química de Drogas VegetalesSIRCARLOS MOLINA RETAMOZOÎncă nu există evaluări
- SodioDocument1 paginăSodiorony-5002469Încă nu există evaluări
- Caso Clinico Leucemia Linfocitica AgudaDocument22 paginiCaso Clinico Leucemia Linfocitica AgudaLesly Del CarpioÎncă nu există evaluări
- Laboratorio N°7 - ProteínasDocument4 paginiLaboratorio N°7 - ProteínaseleazarÎncă nu există evaluări
- Glice Rola DosDocument6 paginiGlice Rola DosLuiz MiguelÎncă nu există evaluări
- Salmonella y Shigella AgarDocument24 paginiSalmonella y Shigella AgarAngie NumpaqueÎncă nu există evaluări
- Laboratorio #3. FagocitosisDocument7 paginiLaboratorio #3. FagocitosisShin Hyun Yoo0% (1)
- Pracctica # 5 Determinación Cuantitativa de Urea en SueroDocument9 paginiPracctica # 5 Determinación Cuantitativa de Urea en SueroPaula Chavez ArellanoÎncă nu există evaluări
- A1C-2 EsDocument7 paginiA1C-2 EsJose Carlos Parras CasasÎncă nu există evaluări
- Práctica 6 EgoDocument7 paginiPráctica 6 EgoSandy MoralesÎncă nu există evaluări
- DIURETICOSssDocument17 paginiDIURETICOSssPablo VallejoÎncă nu există evaluări
- EVIDENCIAS Educacion Fisica - Semana 3docxDocument2 paginiEVIDENCIAS Educacion Fisica - Semana 3docxJonathan MurgaÎncă nu există evaluări
- HT 08 - CetonasDocument2 paginiHT 08 - CetonasJonathan MurgaÎncă nu există evaluări
- EVIDENCIAS DE EDUACIÓN FISICA Semana 03Document1 paginăEVIDENCIAS DE EDUACIÓN FISICA Semana 03Jonathan MurgaÎncă nu există evaluări
- Evidencias 14 y 16 de AbrilDocument2 paginiEvidencias 14 y 16 de AbrilJonathan MurgaÎncă nu există evaluări
- 1ro - CARPETA DE RECUPERACIÓNDocument10 pagini1ro - CARPETA DE RECUPERACIÓNJonathan MurgaÎncă nu există evaluări
- HT 07 - AldehídosDocument2 paginiHT 07 - AldehídosJonathan MurgaÎncă nu există evaluări
- 2° y 3° UNIDAD BIOLOGÍA 5°Document80 pagini2° y 3° UNIDAD BIOLOGÍA 5°Jonathan MurgaÎncă nu există evaluări
- Estructura de TesisDocument1 paginăEstructura de TesisJuan Luis Ledesma GonzalezÎncă nu există evaluări
- Ejercicios Propuestos 4Document2 paginiEjercicios Propuestos 4Jonathan Murga100% (1)
- HT - 11 - AminasDocument2 paginiHT - 11 - AminasmayckolÎncă nu există evaluări
- Solucionario - HT 11 - Aminas PDFDocument2 paginiSolucionario - HT 11 - Aminas PDFAlonso100% (2)
- Factores de ConversiónDocument1 paginăFactores de Conversiónjesusjc0504xÎncă nu există evaluări
- Ejercicios Propuestos-3Document2 paginiEjercicios Propuestos-3Jonathan MurgaÎncă nu există evaluări
- Ejercicios Propuestos 2Document2 paginiEjercicios Propuestos 2Jonathan MurgaÎncă nu există evaluări
- Conoce A Tu Docente - 2019 - 1Document2 paginiConoce A Tu Docente - 2019 - 1Jonathan MurgaÎncă nu există evaluări
- Guia Horas Practica de Campo-2018 - 3Document31 paginiGuia Horas Practica de Campo-2018 - 3Frank MillerÎncă nu există evaluări
- InventariosDocument9 paginiInventariosJonathan MurgaÎncă nu există evaluări
- Ejercicios Propuestos 1Document2 paginiEjercicios Propuestos 1Jonathan MurgaÎncă nu există evaluări
- DIRECCION TACTICAS4okDocument32 paginiDIRECCION TACTICAS4okJonathan MurgaÎncă nu există evaluări
- Invent A Rio Semana 4Document22 paginiInvent A Rio Semana 4Jonathan MurgaÎncă nu există evaluări
- Estructura Capstone Project 19032018Document1 paginăEstructura Capstone Project 19032018Jonathan MurgaÎncă nu există evaluări
- Examen FinalDocument3 paginiExamen FinalJonathan MurgaÎncă nu există evaluări
- Curso - Planeación Agregada - PronósticosDocument15 paginiCurso - Planeación Agregada - PronósticosRichard Oswald Puzza QÎncă nu există evaluări
- La IliadaDocument12 paginiLa IliadaJonathan MurgaÎncă nu există evaluări
- Estructura Cuaderno de CampoDocument1 paginăEstructura Cuaderno de CampoJonathan Murga100% (2)
- Caso PronosticoDocument2 paginiCaso PronosticoJonathan MurgaÎncă nu există evaluări
- Trabajo ParabolasDocument6 paginiTrabajo ParabolasJonathan MurgaÎncă nu există evaluări
- Conociendo La Familia EscolarDocument4 paginiConociendo La Familia EscolarJonathan MurgaÎncă nu există evaluări
- Doc-Th-001 Tablero de Mando de IndicadoresDocument1 paginăDoc-Th-001 Tablero de Mando de IndicadoresJonathan MurgaÎncă nu există evaluări
- TesisDocument88 paginiTesisJonathan MurgaÎncă nu există evaluări
- Fundamentos de Radiologia Dental Erick Whaites PDFDocument488 paginiFundamentos de Radiologia Dental Erick Whaites PDFNaomi Pochon50% (2)
- PostorNoPostDrWalterDevoto2009 EsDocument23 paginiPostorNoPostDrWalterDevoto2009 EsLisseth Dahiana CañasÎncă nu există evaluări
- Clinica Medica IiDocument5 paginiClinica Medica IiMariannyÎncă nu există evaluări
- Zguia de Practicas 2019 de Farmacoquimica Ii FBDocument18 paginiZguia de Practicas 2019 de Farmacoquimica Ii FByoni hernan bances lalupuÎncă nu există evaluări
- Trabajo Final CariesDocument11 paginiTrabajo Final CariesCentro De Atención OdontológicaÎncă nu există evaluări
- LasikDocument6 paginiLasikAlejandra Agüero HaroÎncă nu există evaluări
- El Equipo de Salud en El Ámbito ComunitarioDocument5 paginiEl Equipo de Salud en El Ámbito ComunitarioYanina CastilloÎncă nu există evaluări
- Esquema Psi General Unidad 3 PDFDocument7 paginiEsquema Psi General Unidad 3 PDFPatty100% (1)
- Registro SanitarioDocument13 paginiRegistro SanitarioDavid GonzálezÎncă nu există evaluări
- Neuro Integracion SensorialDocument5 paginiNeuro Integracion SensorialFELIPE AHUMADA AHUMADAÎncă nu există evaluări
- Entrevista PsicopatologicaDocument13 paginiEntrevista PsicopatologicaJunior GavidiaÎncă nu există evaluări
- MARCO TEORICO FarmacognosiaDocument2 paginiMARCO TEORICO FarmacognosiaJimena PinzonÎncă nu există evaluări
- Mof Patologia Clinica 2015Document63 paginiMof Patologia Clinica 2015Nancy BrizuelaÎncă nu există evaluări
- Epidemiologia en EstomatologíaDocument25 paginiEpidemiologia en EstomatologíaSamantaRiveraÎncă nu există evaluări
- Apunte Endocrinología, Diabetes y NutriciónDocument45 paginiApunte Endocrinología, Diabetes y NutriciónMaria Calvo DelgadoÎncă nu există evaluări
- Trastornos Del Calcio, Fosforo Y MagnesioDocument27 paginiTrastornos Del Calcio, Fosforo Y MagnesionadiaÎncă nu există evaluări
- Ley de Curación de HeringDocument3 paginiLey de Curación de HeringMoniDeLanús50% (2)
- UntitledDocument13 paginiUntitledAguirre Arias EfraínÎncă nu există evaluări
- Esquema de VacunaciónDocument1 paginăEsquema de VacunaciónMaría SoledispaÎncă nu există evaluări
- Clomazol 3Document2 paginiClomazol 3Alexandra L Choez100% (1)
- Manual MetapanDocument60 paginiManual MetapanAdolfoRosalesÎncă nu există evaluări
- Trastornos de La Hemostasia SecundariaDocument26 paginiTrastornos de La Hemostasia SecundariaMiguel Corona Perezgrovas100% (1)
- Tema 14 Celador Funciones de VigilanciaDocument4 paginiTema 14 Celador Funciones de VigilanciaTamara Pascual GarridoÎncă nu există evaluări
- Formula Roja HematologiaDocument4 paginiFormula Roja HematologiaMissael JimenezÎncă nu există evaluări
- Guia Clinica Crisis TirotoxicaDocument13 paginiGuia Clinica Crisis TirotoxicaAntonio SaavedraÎncă nu există evaluări
- Actividad Evaluada Unidad IIIDocument16 paginiActividad Evaluada Unidad IIIPunto Las RosasÎncă nu există evaluări
- Glutamato MonosodicoDocument18 paginiGlutamato MonosodicoAaron House100% (1)
- Marco TeoricoDocument7 paginiMarco TeoricoAngelica Castañon0% (1)
- Riesgos de Choque ElectricoDocument25 paginiRiesgos de Choque Electricotiburon53Încă nu există evaluări
- Fisica Transporte AlveolarDocument3 paginiFisica Transporte AlveolarDangelo AlbertoÎncă nu există evaluări