S-ar putea să vă placă și
- Diseases of the Liver and Biliary TreeDe la EverandDiseases of the Liver and Biliary TreeAnnarosa FloreaniÎncă nu există evaluări
- Tracheo Esophageal FistulaDocument6 paginiTracheo Esophageal Fistulablast2111Încă nu există evaluări
- BaldwinDocument6 paginiBaldwinalexafarrenÎncă nu există evaluări
- Fanaroff Anormalidades GastrointestinalesDocument28 paginiFanaroff Anormalidades GastrointestinalesDome HerreraÎncă nu există evaluări
- Case Report Esophageal AtresiaDocument3 paginiCase Report Esophageal Atresiaadriani lawrenciaÎncă nu există evaluări
- Congenital Anomalies of The Esophagus PDFDocument3 paginiCongenital Anomalies of The Esophagus PDFSpecialName100% (1)
- Esophageal Atresia: Future Directions For Research On The Digestive TractDocument7 paginiEsophageal Atresia: Future Directions For Research On The Digestive TractindadzilarsyÎncă nu există evaluări
- Esophageal AtresiaDocument21 paginiEsophageal AtresiaOktania Putri Kusnawan100% (2)
- Congenital AnomaliesDocument10 paginiCongenital Anomaliesربيع ضياء ربيعÎncă nu există evaluări
- Oesophagealatresia 120920081900 Phpapp01Document37 paginiOesophagealatresia 120920081900 Phpapp01Mulugeta AbenehÎncă nu există evaluări
- Semin Fetal Neonatal Med 2011 Jun 16 (3) 164 Wall Defects PDFDocument9 paginiSemin Fetal Neonatal Med 2011 Jun 16 (3) 164 Wall Defects PDFrestoe agustinÎncă nu există evaluări
- Management of Neonates With Oesophageal Atresia and TR - 2022 - Early Human DeveDocument6 paginiManagement of Neonates With Oesophageal Atresia and TR - 2022 - Early Human DeveJUAN HIMMEL ESPINOZA ANDIAÎncă nu există evaluări
- Gastroschisis Omphalocele PDFDocument5 paginiGastroschisis Omphalocele PDFFariz Eka SetiawanÎncă nu există evaluări
- Abdomina Wall Defects 2Document12 paginiAbdomina Wall Defects 2pamellapesseÎncă nu există evaluări
- Anaesthesia For Prune Belly Syndrome: Syndromic Vignettes in AnaesthesiaDocument2 paginiAnaesthesia For Prune Belly Syndrome: Syndromic Vignettes in AnaesthesiaIonut BaraÎncă nu există evaluări
- World Surgery: DuodenumDocument2 paginiWorld Surgery: DuodenumBeladiena Citra SiregarÎncă nu există evaluări
- Current Knowledge On Esophageal Atresia: Go ToDocument21 paginiCurrent Knowledge On Esophageal Atresia: Go TonurulÎncă nu există evaluări
- Tracheoesophageal Atresia: Mrs - Smitha.M Associate Professor Vijaya College of Nursing KottarakkaraDocument10 paginiTracheoesophageal Atresia: Mrs - Smitha.M Associate Professor Vijaya College of Nursing KottarakkarakrishnasreeÎncă nu există evaluări
- Acalasia PDFDocument11 paginiAcalasia PDFFrancisco EspinozaÎncă nu există evaluări
- Apple Peel Small Bowel, A Review of Four Cases: Surgical and Radiographic AspectsDocument8 paginiApple Peel Small Bowel, A Review of Four Cases: Surgical and Radiographic AspectsraecmyÎncă nu există evaluări
- Shalita Dastamuar Pediatric Surgery Subdivision, Surgery Department Mohammad Hoesin HospitalDocument18 paginiShalita Dastamuar Pediatric Surgery Subdivision, Surgery Department Mohammad Hoesin HospitalSoni TfacÎncă nu există evaluări
- Journal AsliDocument4 paginiJournal Aslidesi tethunÎncă nu există evaluări
- Herman 2012Document20 paginiHerman 2012dewaprasatyaÎncă nu există evaluări
- Atresia de Esófago Long Gap. Definición y Conducta Quirúrgica ModernaDocument8 paginiAtresia de Esófago Long Gap. Definición y Conducta Quirúrgica ModernaGUDELIA JIMENEZ SANCHEZÎncă nu există evaluări
- Intestinal Atresia: A Case ReportDocument3 paginiIntestinal Atresia: A Case ReportOvamelia JulioÎncă nu există evaluări
- Surgical Treatment of Gastrointestinal Motility DisordersDocument47 paginiSurgical Treatment of Gastrointestinal Motility DisordersBolivar IseaÎncă nu există evaluări
- Embryology of Oesophageal Atresia: Adonis S. Ioannides, Andrew J. CoppDocument10 paginiEmbryology of Oesophageal Atresia: Adonis S. Ioannides, Andrew J. CoppXimenita GhilardiÎncă nu există evaluări
- Pre Fumo 2014Document12 paginiPre Fumo 2014LiaApprilia Kartinii Cupcupcuap ElvenadoÎncă nu există evaluări
- Gastro SCH Is IsDocument13 paginiGastro SCH Is IsferoÎncă nu există evaluări
- Airway AnomaliesDocument11 paginiAirway AnomaliesDariki CampbellÎncă nu există evaluări
- Sabitson - Appendiks EngDocument17 paginiSabitson - Appendiks Engzeek powerÎncă nu există evaluări
- Medip, IJCP-1422 CDocument3 paginiMedip, IJCP-1422 CdsagemaverickÎncă nu există evaluări
- PSU Vol 06, 1996Document5 paginiPSU Vol 06, 1996rajarshikÎncă nu există evaluări
- Prune Belly SyndromeDocument39 paginiPrune Belly SyndromeHudaÎncă nu există evaluări
- Neonatal Bowel Obstruction: David Juang,, Charles L. SnyderDocument27 paginiNeonatal Bowel Obstruction: David Juang,, Charles L. SnyderNaty BmÎncă nu există evaluări
- Summary and Conclusion Esophageal Atresia (Or Oesophageal AtresiaDocument3 paginiSummary and Conclusion Esophageal Atresia (Or Oesophageal AtresiaSanket TelangÎncă nu există evaluări
- Journal of Rare Diseases MARDocument13 paginiJournal of Rare Diseases MARDaiiny DelgadoÎncă nu există evaluări
- Kayatsha - Gastroschisis and Omphalocele A Case ReportDocument4 paginiKayatsha - Gastroschisis and Omphalocele A Case ReportAffannul HakimÎncă nu există evaluări
- 1940 12050 1 PBDocument7 pagini1940 12050 1 PBmano mayÎncă nu există evaluări
- EsofagusDocument7 paginiEsofagusWillie VanegasÎncă nu există evaluări
- Esophageal Atresia: DR - Ibrahim AlsbouDocument21 paginiEsophageal Atresia: DR - Ibrahim Alsbouraed faisalÎncă nu există evaluări
- Achalasia: An Overview of Diagnosis and Treatment: ReviewDocument7 paginiAchalasia: An Overview of Diagnosis and Treatment: ReviewJessica GintingÎncă nu există evaluări
- Why So Many Patients With Dysphagia Have Normal Esophageal Function TestingDocument13 paginiWhy So Many Patients With Dysphagia Have Normal Esophageal Function Testingdraanalordonezv1991Încă nu există evaluări
- Congenital AnomaliesDocument22 paginiCongenital Anomaliesjessy100% (1)
- Intussusception in Children - A Clinical ReviewDocument8 paginiIntussusception in Children - A Clinical Reviewjohanrubiano6Încă nu există evaluări
- Trilogy of Foregut Midgut and Hindgut Atresias PreDocument5 paginiTrilogy of Foregut Midgut and Hindgut Atresias PreEriekafebriayana RÎncă nu există evaluări
- Nihms 1657970Document20 paginiNihms 1657970mchojnacki81Încă nu există evaluări
- Imaging Patients With Acute Abdominal PainDocument16 paginiImaging Patients With Acute Abdominal PainNaelul IzahÎncă nu există evaluări
- Esophageal Atresia and Tracheoesophageal FistulaDocument28 paginiEsophageal Atresia and Tracheoesophageal FistulaarifÎncă nu există evaluări
- Atresia EsophagusDocument6 paginiAtresia EsophagusPPN38 UNPADÎncă nu există evaluări
- Pediatric Laryngotracheal Stenosis and Airway Reconstruction: A Review of Voice Outcomes, Assessment, and Treatment IssuesDocument11 paginiPediatric Laryngotracheal Stenosis and Airway Reconstruction: A Review of Voice Outcomes, Assessment, and Treatment Issuescanndy202Încă nu există evaluări
- Case Studies: OEIS ComplexDocument6 paginiCase Studies: OEIS ComplexHuzayval AchmadÎncă nu există evaluări
- AtresiaDocument5 paginiAtresiakirtiÎncă nu există evaluări
- Open Cervical Surgery For Congenital H-Type Tracheoesophageal Fistulae - TOFDocument8 paginiOpen Cervical Surgery For Congenital H-Type Tracheoesophageal Fistulae - TOFHussein AhmedÎncă nu există evaluări
- Bedah Anak: Kelainan Kongenital Gastrointestinal Anak Kedaruratan AnakDocument52 paginiBedah Anak: Kelainan Kongenital Gastrointestinal Anak Kedaruratan AnakalifÎncă nu există evaluări
- Soleimani 2007Document9 paginiSoleimani 2007ceciliaÎncă nu există evaluări
- Life-Saving Esophageal Intubation in Neonate With Undiagnosed Tracheal Agenesis (A & A Case Reports, Vol. 9, Issue 1) (2017) DGDFGDocument2 paginiLife-Saving Esophageal Intubation in Neonate With Undiagnosed Tracheal Agenesis (A & A Case Reports, Vol. 9, Issue 1) (2017) DGDFGMahdi WashahaÎncă nu există evaluări
- Screenshot 2022-05-30 at 9.43.21 PMDocument23 paginiScreenshot 2022-05-30 at 9.43.21 PMAZÎncă nu există evaluări
- OmphaloceleDocument23 paginiOmphaloceleFred PupeÎncă nu există evaluări
- Branski (2011)Document7 paginiBranski (2011)TommysÎncă nu există evaluări
- Lalkhen 2011Document5 paginiLalkhen 2011TommysÎncă nu există evaluări
- Surgical Management of Intracerebral HemorrhageDocument10 paginiSurgical Management of Intracerebral HemorrhageTommysÎncă nu există evaluări
- CR - RamirezDocument4 paginiCR - RamirezTommysÎncă nu există evaluări
- AJCC Breast Cancer Staging SystemDocument50 paginiAJCC Breast Cancer Staging SystemLucan MihaelaÎncă nu există evaluări
- The Role of Obesity and Related Metabolic Disturbances in Cancers ofDocument18 paginiThe Role of Obesity and Related Metabolic Disturbances in Cancers ofTommysÎncă nu există evaluări
- Mun (2001)Document10 paginiMun (2001)TommysÎncă nu există evaluări
- Appleton (2006)Document6 paginiAppleton (2006)TommysÎncă nu există evaluări
- Chronic InflammationDocument24 paginiChronic InflammationTommys100% (1)
- Periprosthetic Fractures in Total Knee ArthroplastyDocument6 paginiPeriprosthetic Fractures in Total Knee ArthroplastyTommysÎncă nu există evaluări
- Andrew (2015)Document13 paginiAndrew (2015)TommysÎncă nu există evaluări
- Festing 2002Document15 paginiFesting 2002TommysÎncă nu există evaluări
- Ehlinger (2010)Document6 paginiEhlinger (2010)TommysÎncă nu există evaluări
- Service Evaluation, Audit and Research: What Is The Difference?Document2 paginiService Evaluation, Audit and Research: What Is The Difference?TommysÎncă nu există evaluări
- Fredholm1982 Chronic Caffeine in RatsDocument4 paginiFredholm1982 Chronic Caffeine in RatsTommysÎncă nu există evaluări
- Mte (2014)Document14 paginiMte (2014)TommysÎncă nu există evaluări
- WCSQ Full PaperDocument15 paginiWCSQ Full PaperTommysÎncă nu există evaluări
- Ni Hms 79693Document17 paginiNi Hms 79693TommysÎncă nu există evaluări
- Surviving Sepsis Campaign: International Guidelines For Management of Severe Sepsis and Septic Shock: 2016Document56 paginiSurviving Sepsis Campaign: International Guidelines For Management of Severe Sepsis and Septic Shock: 2016TommysÎncă nu există evaluări
- Jurnal Trauma KepalaDocument7 paginiJurnal Trauma KepalaTommysÎncă nu există evaluări
- Branski (2011)Document7 paginiBranski (2011)TommysÎncă nu există evaluări
- Green Hal GHDocument12 paginiGreen Hal GHTommysÎncă nu există evaluări
- DDDT 8 1923 PDFDocument6 paginiDDDT 8 1923 PDFTommysÎncă nu există evaluări
- Skin Flap Dr. BrotoDocument17 paginiSkin Flap Dr. BrotoTommysÎncă nu există evaluări
- Emergency Management of Severe Burns Manual: February 2012Document3 paginiEmergency Management of Severe Burns Manual: February 2012TommysÎncă nu există evaluări
- CaffeineDocument9 paginiCaffeineTommysÎncă nu există evaluări
- Coffee StatisticsDocument5 paginiCoffee StatisticsTommysÎncă nu există evaluări
- Yuwono (2014) - Coffee PowderDocument5 paginiYuwono (2014) - Coffee PowderTommysÎncă nu există evaluări
- Yuwono (2017) - Coffee ReviewDocument3 paginiYuwono (2017) - Coffee ReviewTommysÎncă nu există evaluări
- Daewoo 710B PDFDocument59 paginiDaewoo 710B PDFbgmentÎncă nu există evaluări
- Ti05001 PDFDocument1 paginăTi05001 PDFanggieÎncă nu există evaluări
- Research Group 3 11abmb1Document32 paginiResearch Group 3 11abmb1arianeÎncă nu există evaluări
- Backwards Design - Jessica W Maddison CDocument20 paginiBackwards Design - Jessica W Maddison Capi-451306299100% (1)
- Consent Form: Republic of The Philippines Province of - Municipality ofDocument1 paginăConsent Form: Republic of The Philippines Province of - Municipality ofLucette Legaspi EstrellaÎncă nu există evaluări
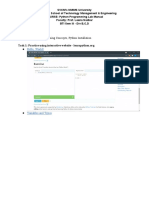
- Week - 2 Lab - 1 - Part I Lab Aim: Basic Programming Concepts, Python InstallationDocument13 paginiWeek - 2 Lab - 1 - Part I Lab Aim: Basic Programming Concepts, Python InstallationSahil Shah100% (1)
- DS Important QuestionsDocument15 paginiDS Important QuestionsLavanya JÎncă nu există evaluări
- Galman V PamaranDocument7 paginiGalman V PamaranChow Momville EstimoÎncă nu există evaluări
- Advocacy Firm Business Plan by SlidesgoDocument40 paginiAdvocacy Firm Business Plan by SlidesgoirinaÎncă nu există evaluări
- Sales Forecast Template DownloadDocument9 paginiSales Forecast Template DownloadAshokÎncă nu există evaluări
- B. Inggris X - 7Document8 paginiB. Inggris X - 7KabardiantoÎncă nu există evaluări
- Group 2 ITI Consensus Report: Prosthodontics and Implant DentistryDocument9 paginiGroup 2 ITI Consensus Report: Prosthodontics and Implant DentistryEsme ValenciaÎncă nu există evaluări
- MDI - Good Fellas - ScriptDocument20 paginiMDI - Good Fellas - ScriptRahulSamaddarÎncă nu există evaluări
- Sheltered 2 Item Recycle ListDocument5 paginiSheltered 2 Item Recycle ListRachel GÎncă nu există evaluări
- Fortigate Firewall Version 4 OSDocument122 paginiFortigate Firewall Version 4 OSSam Mani Jacob DÎncă nu există evaluări
- Generalized Class of Sakaguchi Functions in Conic Region: Saritha. G. P, Fuad. S. Al Sarari, S. LathaDocument5 paginiGeneralized Class of Sakaguchi Functions in Conic Region: Saritha. G. P, Fuad. S. Al Sarari, S. LathaerpublicationÎncă nu există evaluări
- Spare Part PhilosophyDocument27 paginiSpare Part Philosophyavaisharma50% (2)
- European Construction Sector Observatory: Country Profile MaltaDocument40 paginiEuropean Construction Sector Observatory: Country Profile MaltaRainbootÎncă nu există evaluări
- BSH 7005-15Document129 paginiBSH 7005-15Mark InnesÎncă nu există evaluări
- Jpedal ManualDocument20 paginiJpedal ManualDamián DávilaÎncă nu există evaluări
- Republic of The Philippines Division of Bohol Department of Education Region VII, Central VisayasDocument6 paginiRepublic of The Philippines Division of Bohol Department of Education Region VII, Central VisayasJOHN MC RAE RACINESÎncă nu există evaluări
- RECYFIX STANDARD 100 Tipe 010 MW - C250Document2 paginiRECYFIX STANDARD 100 Tipe 010 MW - C250Dadang KurniaÎncă nu există evaluări
- Designing and Drawing PropellerDocument4 paginiDesigning and Drawing Propellercumpio425428100% (1)
- Presenters: Horace M. Estrella Jay Mart A. Lazana Princess Camille R. HipolitoDocument23 paginiPresenters: Horace M. Estrella Jay Mart A. Lazana Princess Camille R. HipolitoHorace EstrellaÎncă nu există evaluări
- Lesson 1 Q3 Figure Life DrawingDocument10 paginiLesson 1 Q3 Figure Life DrawingCAHAPÎncă nu există evaluări
- Concrete Specification (BS8500)Document3 paginiConcrete Specification (BS8500)teh100% (1)
- Benjamin Franklin - The Indian Treaties (1938)Document450 paginiBenjamin Franklin - The Indian Treaties (1938)Spiritu SanctoÎncă nu există evaluări
- EP001 LifeCoachSchoolTranscriptDocument13 paginiEP001 LifeCoachSchoolTranscriptVan GuedesÎncă nu există evaluări
- Rishika Reddy Art Integrated ActivityDocument11 paginiRishika Reddy Art Integrated ActivityRishika ReddyÎncă nu există evaluări
- Raksha Mantralaya Ministry of DefenceDocument16 paginiRaksha Mantralaya Ministry of Defencesubhasmita sahuÎncă nu există evaluări