S-ar putea să vă placă și
- Ingeniería de procesos siderúrgicos: La experiencia de AHMSADe la EverandIngeniería de procesos siderúrgicos: La experiencia de AHMSAÎncă nu există evaluări
- Análisis de Cobre Por Espectroscopía de Absorción Atómica - FORO TEMATICO DEL TR1 PDFDocument6 paginiAnálisis de Cobre Por Espectroscopía de Absorción Atómica - FORO TEMATICO DEL TR1 PDFRuth TenorioÎncă nu există evaluări
- Horno de Grafito Espectroscopia de Absorcion AtomicaDocument9 paginiHorno de Grafito Espectroscopia de Absorcion AtomicaEduardo LuqueÎncă nu există evaluări
- Determinación de Hierro en Lentejas Por Absorción AtómicaDocument13 paginiDeterminación de Hierro en Lentejas Por Absorción AtómicaDalila Isabel Falcón Paz100% (1)
- 6.2. Espectrofotometría de Absorción AtómicaDocument8 pagini6.2. Espectrofotometría de Absorción AtómicaMaximiliano DelahigueraÎncă nu există evaluări
- Trabajo Grupal 1 AIDocument17 paginiTrabajo Grupal 1 AIJorge Ricardo AlcivarÎncă nu există evaluări
- Absorcion Atomica2Document19 paginiAbsorcion Atomica2Veronica NepoÎncă nu există evaluări
- Determinación de Fe Por Absorción AtómicaDocument12 paginiDeterminación de Fe Por Absorción AtómicaReynaldo Ponte RamirezÎncă nu există evaluări
- Nte Inen 2682-Espectrometria-Absorcion AtomicaDocument15 paginiNte Inen 2682-Espectrometria-Absorcion Atomicashirley changÎncă nu există evaluări
- Espectrometria AtomicaDocument6 paginiEspectrometria AtomicaJOCELYN MARINÎncă nu există evaluări
- Sistema de Digestión Asistida Por Microondas e ICP-MS PDFDocument10 paginiSistema de Digestión Asistida Por Microondas e ICP-MS PDFAnonymous tOZnJYUÎncă nu există evaluări
- Análisis de Mercurio Por Absorción AtomicaDocument21 paginiAnálisis de Mercurio Por Absorción AtomicaKelvin Briceño SalazarÎncă nu există evaluări
- Diapositivas KarenDocument13 paginiDiapositivas KarenKaren Margarita Ramos PerezÎncă nu există evaluări
- Martínez - Técnicas de Generación de Vapor Acopladas A Espectrofotometría de Absorción AtómicaDocument8 paginiMartínez - Técnicas de Generación de Vapor Acopladas A Espectrofotometría de Absorción AtómicaMiguel Eduardo Velázquez HernándezÎncă nu există evaluări
- Quimicaa 12Document16 paginiQuimicaa 12Paola PerezÎncă nu există evaluări
- CVAASDocument4 paginiCVAASjose perezÎncă nu există evaluări
- Instrumental P 8 e 6Document10 paginiInstrumental P 8 e 6Alejandra HernandezÎncă nu există evaluări
- Etanol Cromatografía de GasesDocument16 paginiEtanol Cromatografía de GasesLeslie AlmanzaÎncă nu există evaluări
- Concentracion de GasesDocument6 paginiConcentracion de GasesAurora Castro CastilloÎncă nu există evaluări
- Protocolo para La Determinacion de HierroDocument8 paginiProtocolo para La Determinacion de HierroFabían Díaz Jhordan JhampierdÎncă nu există evaluări
- Reporte de Práctica No.5Document7 paginiReporte de Práctica No.5andrea.gutierrez0542Încă nu există evaluări
- Presentación Espectroscopía AtómicaDocument53 paginiPresentación Espectroscopía AtómicaAna Florencia NadalÎncă nu există evaluări
- Plata y OroDocument29 paginiPlata y OroRoy Juan Mendoza Palma100% (1)
- Horno de Grafito PDFDocument20 paginiHorno de Grafito PDFMarita M. Orbegoso100% (1)
- Inf 9 CuantiDocument30 paginiInf 9 CuantipomposoÎncă nu există evaluări
- Informe 2 Ana InstruhhhDocument19 paginiInforme 2 Ana InstruhhhNilton Cj100% (1)
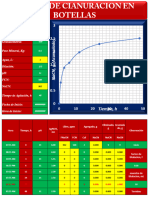
- Informe de Cianuracion en BotellaDocument32 paginiInforme de Cianuracion en Botellajhoni88% (8)
- Análisis de Magnesio Por Absorción AtómicaDocument24 paginiAnálisis de Magnesio Por Absorción AtómicaJosue Garcia Yaranga100% (3)
- Absorcion Atomica ExposicionDocument72 paginiAbsorcion Atomica ExposicionBranco Figueroa AyalaÎncă nu există evaluări
- UOP Mercury Removal From-Natural Gas-And Liquid-Stream-1Document9 paginiUOP Mercury Removal From-Natural Gas-And Liquid-Stream-1Andres MonsalveÎncă nu există evaluări
- Análisis de Sodio y Potasio Por Eaa Lectura Directa en AguasDocument10 paginiAnálisis de Sodio y Potasio Por Eaa Lectura Directa en AguasRosalynNeciosupRamosÎncă nu există evaluări
- 2do ANÁLISIS DE MINERALES POR ESPECTROFOTOMETRÍA DE ABSORCIÓN ATÓMICADocument5 pagini2do ANÁLISIS DE MINERALES POR ESPECTROFOTOMETRÍA DE ABSORCIÓN ATÓMICAGRACIELA QUISPE HUAMANÎncă nu există evaluări
- Métodos de Análisis de Metales Pesados en Agua TerminadoDocument20 paginiMétodos de Análisis de Metales Pesados en Agua TerminadoPatricia Pereda SimoniÎncă nu există evaluări
- Practica 5Document16 paginiPractica 5Jair Paredes Ramirez60% (5)
- Fotometria de LlamaDocument5 paginiFotometria de LlamaDiannys RodriguezÎncă nu există evaluări
- GRUPO 02 - Fotometria de Llama (FINAL)Document35 paginiGRUPO 02 - Fotometria de Llama (FINAL)Norka Rosmery Asqui CabreraÎncă nu există evaluări
- Informe Absorción Atómica-2Document5 paginiInforme Absorción Atómica-2karo muñozÎncă nu există evaluări
- Lab #8 EAA Qca Anal.Document6 paginiLab #8 EAA Qca Anal.Grii Rodriguez100% (1)
- Determinacion de ZincDocument5 paginiDeterminacion de ZincrubyÎncă nu există evaluări
- Especrroscopia de AbsorcionDocument3 paginiEspecrroscopia de AbsorcionNestor SanchezÎncă nu există evaluări
- 2ap35 PDFDocument1 pagină2ap35 PDFEduÎncă nu există evaluări
- Instrumentación para La Cromatografía de GasesDocument24 paginiInstrumentación para La Cromatografía de Gasescrisina Arteaga100% (1)
- Capitulo 2Document47 paginiCapitulo 2Miguel Reynaldo CabreraÎncă nu există evaluări
- Química AnalíticaDocument28 paginiQuímica AnalíticaOLGA VEGAÎncă nu există evaluări
- Guialab 8Document5 paginiGuialab 8Liga ChilenaÎncă nu există evaluări
- DETERMINACIÓN DE COBRE Y ZINC EN CABELLO POR ESPECTROFOTOMETRÍA DE ABSORCIÓN ATÓMICA 11-11-13 (10.37 PM)Document6 paginiDETERMINACIÓN DE COBRE Y ZINC EN CABELLO POR ESPECTROFOTOMETRÍA DE ABSORCIÓN ATÓMICA 11-11-13 (10.37 PM)Andrea BejaranoÎncă nu există evaluări
- Resumen de Cromatografia de GasesDocument13 paginiResumen de Cromatografia de GasesDan MatthewsÎncă nu există evaluări
- Informe Arsénico GDocument18 paginiInforme Arsénico GEvelyn AguilarÎncă nu există evaluări
- TPL 4 - Abs AtómicaDocument13 paginiTPL 4 - Abs AtómicaEdupanda m100% (1)
- Absorcion AtómicaDocument7 paginiAbsorcion AtómicaFrank F Vargas BÎncă nu există evaluări
- Aqi 13 2020-I UnmsmDocument27 paginiAqi 13 2020-I UnmsmC'Denisse PalominoÎncă nu există evaluări
- DETERMINACIÓN DE ZN EN CONSERVAS DE PESCADO POR ABSORCION ATÓMICA EN LLAMADocument6 paginiDETERMINACIÓN DE ZN EN CONSERVAS DE PESCADO POR ABSORCION ATÓMICA EN LLAMAValeriaÎncă nu există evaluări
- Cromatografia de GasesDocument7 paginiCromatografia de Gasesjose manuel arancibia barahonaÎncă nu există evaluări
- Grupo 02 - Ingenieria AmbientalDocument35 paginiGrupo 02 - Ingenieria AmbientalNorka Rosmery Asqui CabreraÎncă nu există evaluări
- Expo Analisis Determinacion Por EspectrosDocument23 paginiExpo Analisis Determinacion Por EspectrosCitlali MartínezÎncă nu există evaluări
- Deber 4Document7 paginiDeber 4Gabriel GarciaÎncă nu există evaluări
- Presentación Cromatografía de GasesDocument40 paginiPresentación Cromatografía de GasesyaxemÎncă nu există evaluări
- E. Absorción AtómicaDocument21 paginiE. Absorción AtómicaGiselle CaoÎncă nu există evaluări
- Preguntas de Repaso 1ra SemanaDocument10 paginiPreguntas de Repaso 1ra SemanaJhonathan Cutipa QuispeÎncă nu există evaluări
- LadrilloDocument8 paginiLadrilloBartholomew Beauregard VanderbiltÎncă nu există evaluări
- Monografia Del Caolin y Arcilla Quimica InorganicaDocument60 paginiMonografia Del Caolin y Arcilla Quimica InorganicaJhonathan Cutipa QuispeÎncă nu există evaluări
- Ley Del Código de Ética de La Función Pública-1 PDFDocument4 paginiLey Del Código de Ética de La Función Pública-1 PDFRaul Rojas HÎncă nu există evaluări
- Tipos de Ladrillos para ConstrucciónDocument3 paginiTipos de Ladrillos para ConstrucciónJhonathan Cutipa QuispeÎncă nu există evaluări
- Proyecto de Ladrillos 1Document69 paginiProyecto de Ladrillos 1Jhonathan Cutipa QuispeÎncă nu există evaluări
- Introduccion A La Mecanica Cuantica PDFDocument276 paginiIntroduccion A La Mecanica Cuantica PDFJhon F. RodriguezÎncă nu există evaluări
- Teoria Hecha Por Ceballos PDFDocument115 paginiTeoria Hecha Por Ceballos PDFJorge Condori VilcaÎncă nu există evaluări
- 21 DaysDocument16 pagini21 DaysJhonathan Cutipa QuispeÎncă nu există evaluări
- Informe Visita JuliacaDocument11 paginiInforme Visita JuliacaJhonathan Cutipa QuispeÎncă nu există evaluări
- Laboratorio #12Document7 paginiLaboratorio #12Jhonathan Cutipa QuispeÎncă nu există evaluări
- Extraccion de Aceites Esenciales de Eucalipto Por Destilacion de Arrastre de VaporDocument5 paginiExtraccion de Aceites Esenciales de Eucalipto Por Destilacion de Arrastre de VaporJhonathan Cutipa QuispeÎncă nu există evaluări
- Reconocimientos de Tipos de Fluido1 Feno0menos ImprimrDocument12 paginiReconocimientos de Tipos de Fluido1 Feno0menos ImprimrJhonathan Cutipa QuispeÎncă nu există evaluări
- Determinacion Del Tipo de Flujo ImprimiDocument17 paginiDeterminacion Del Tipo de Flujo ImprimiJhonathan Cutipa QuispeÎncă nu există evaluări
- AlvaroDocument2 paginiAlvaroJhonathan Cutipa QuispeÎncă nu există evaluări
- Aguas Informe 2 AlcalinidadDocument12 paginiAguas Informe 2 AlcalinidadJhonathan Cutipa QuispeÎncă nu există evaluări
- El Pensamiento Andino I EdiciónDocument9 paginiEl Pensamiento Andino I EdiciónJhonathan Cutipa QuispeÎncă nu există evaluări
- UNIVERSIDAD NACIONAL DEL Altiplano FicoDocument10 paginiUNIVERSIDAD NACIONAL DEL Altiplano FicoJhonathan Cutipa QuispeÎncă nu există evaluări
- PF PDFDocument27 paginiPF PDFManuel M Anders CasillasÎncă nu există evaluări
- Directorio FuncionariosDocument19 paginiDirectorio FuncionariosJhonathan Cutipa QuispeÎncă nu există evaluări
- Capítulo 3Document45 paginiCapítulo 3Jhonathan Cutipa QuispeÎncă nu există evaluări
- HttpsDocument4 paginiHttpsJhonathan Cutipa QuispeÎncă nu există evaluări
- LaboratorioDocument27 paginiLaboratorioJhonathan Cutipa QuispeÎncă nu există evaluări
- Ecuaciones DiferencialesDocument2 paginiEcuaciones DiferencialesJhonathan Cutipa QuispeÎncă nu există evaluări
- FISICO QUIMICA Problemas Resueltos de Gases IdealesDocument6 paginiFISICO QUIMICA Problemas Resueltos de Gases IdealesSalvador Mantilla83% (6)
- Tes Is FinalDocument205 paginiTes Is FinalAdolfo Gomez MoralesÎncă nu există evaluări
- Articulo CientificoDocument3 paginiArticulo CientificoJhonathan Cutipa QuispeÎncă nu există evaluări
- Guiade 1Document6 paginiGuiade 1Santiago Ortiz PérezÎncă nu există evaluări
- Universidad Nacional Del Altiplano La MuñaDocument17 paginiUniversidad Nacional Del Altiplano La MuñaJhonathan Cutipa QuispeÎncă nu există evaluări
- Atomización de LlamaDocument23 paginiAtomización de LlamaJhonathan Cutipa QuispeÎncă nu există evaluări
- 02 Etica Transparencia YBuen GobiernoDocument15 pagini02 Etica Transparencia YBuen GobiernoJuan MontesinosÎncă nu există evaluări
- Anexo "F" Certificado Médico (Para Civiles Que Participan en El Proceso de Admisión)Document2 paginiAnexo "F" Certificado Médico (Para Civiles Que Participan en El Proceso de Admisión)Mafer Vazquez100% (1)
- Embriología Semana 3-Práctica FetalDocument12 paginiEmbriología Semana 3-Práctica FetalIzamar Llaure FloresÎncă nu există evaluări
- Intoxicación Por OrganocloradosDocument8 paginiIntoxicación Por OrganocloradosDiego Ƨanchez MarmolejoÎncă nu există evaluări
- Evaluacion de Recuperacion 5°Document10 paginiEvaluacion de Recuperacion 5°Pathie JocelinÎncă nu există evaluări
- Determinación Toxicológica de Cocaína en Humor Vítreo Mmf161fDocument8 paginiDeterminación Toxicológica de Cocaína en Humor Vítreo Mmf161fantonio garridoÎncă nu există evaluări
- Dialnet LaInvestigacionEnElLugarDeLosHechos 4767889 PDFDocument8 paginiDialnet LaInvestigacionEnElLugarDeLosHechos 4767889 PDFMereyin HurtadoÎncă nu există evaluări
- Prueba Anti CCP IChroma IIDocument3 paginiPrueba Anti CCP IChroma IIAda Belen AlvarezÎncă nu există evaluări
- La Condición Física en La Construcción Histórica de La Educación FísicaDocument2 paginiLa Condición Física en La Construcción Histórica de La Educación FísicaRolando GtmeroÎncă nu există evaluări
- Mecanismos de Absorción - 202265 - 202631Document2 paginiMecanismos de Absorción - 202265 - 202631Danilo OtoÎncă nu există evaluări
- Obesidad MaslowUCVDocument15 paginiObesidad MaslowUCVMariaÎncă nu există evaluări
- Jerarquia de Las Necesidades - Abraham MaslowDocument5 paginiJerarquia de Las Necesidades - Abraham MaslowCarlos OsoriioÎncă nu există evaluări
- Ausentismo LaboralDocument21 paginiAusentismo LaboralIngrid Jineth MontoyaÎncă nu există evaluări
- Seminario Obstrucción UrinariaDocument19 paginiSeminario Obstrucción UrinariaGiselle C. Ramos FlautesÎncă nu există evaluări
- COVID19Document59 paginiCOVID19Jesús Abásolo MuñozÎncă nu există evaluări
- Act 1 Linea TiempoDocument3 paginiAct 1 Linea Tiempojose alberto camacho cortesÎncă nu există evaluări
- Morales Moreno Karla CreedDocument4 paginiMorales Moreno Karla CreedCristina Cumpa chungaÎncă nu există evaluări
- Revista de Psiquiatría LatinoamericanaDocument57 paginiRevista de Psiquiatría LatinoamericanaMariana LópezÎncă nu există evaluări
- 02 04Texto2-2-EN ESThe11minuteworkoutDocument2 pagini02 04Texto2-2-EN ESThe11minuteworkoutKARLA JOSE MENDEZ CRUZÎncă nu există evaluări
- Trabajo Final ForosDocument20 paginiTrabajo Final ForosSol AyléÎncă nu există evaluări
- Estudio Del Financiamiento de La SaludDocument376 paginiEstudio Del Financiamiento de La SaludMia IsabellaÎncă nu există evaluări
- COMINODocument5 paginiCOMINOAntonio samaelÎncă nu există evaluări
- Sindrome MeningeoDocument24 paginiSindrome MeningeoDiego Pantoja RamosÎncă nu există evaluări
- LesionologiaDocument5 paginiLesionologiaJhon Brayan Perez CruzattÎncă nu există evaluări
- Informe de Apoyo Sve-DmeDocument13 paginiInforme de Apoyo Sve-DmeriveravarelaÎncă nu există evaluări
- Exfoliación Corp C16Document15 paginiExfoliación Corp C16Kenny Montes Puntillo100% (1)
- Octeto DFRONZO - En.esDocument23 paginiOcteto DFRONZO - En.esAinara SanchezÎncă nu există evaluări
- MSDS - Cemento Nacional - Tipo ICo 2024Document7 paginiMSDS - Cemento Nacional - Tipo ICo 2024OscarVelezmoroÎncă nu există evaluări
- Gestion Ambiental en La Construccion-Parcial IDocument5 paginiGestion Ambiental en La Construccion-Parcial IDeiler ReyesÎncă nu există evaluări
- Oráculo de Gaia Mazo y GuiaDocument97 paginiOráculo de Gaia Mazo y GuiaFlor de Kardo84% (56)