S-ar putea să vă placă și
- SOP Equipment ValidationDocument15 paginiSOP Equipment Validationfarjana100% (7)
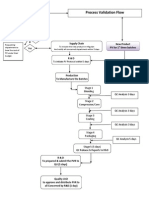
- Flow Chart Process ValidationDocument1 paginăFlow Chart Process Validationmasthan6yÎncă nu există evaluări
- VMP Guide PDFDocument6 paginiVMP Guide PDFsitimunawarohÎncă nu există evaluări
- Validation Summary Report Template PDFDocument12 paginiValidation Summary Report Template PDFAnonymous OreX6RrRP100% (1)
- VAL 080 Validation Master Plan Sample PDFDocument3 paginiVAL 080 Validation Master Plan Sample PDFsiva sankar100% (1)
- Equipment Logbook 2 2Document7 paginiEquipment Logbook 2 2Belazouz BoualemÎncă nu există evaluări
- A WHO GuideDocument100 paginiA WHO Guidej.k.kumar92% (13)
- Q Pharma Quality ManualDocument32 paginiQ Pharma Quality Manualsappz354544883% (6)
- Quality eCTD SubmissionsDocument7 paginiQuality eCTD SubmissionsRambabu komati - QA100% (6)
- Tim Fields Master Validation PlanDocument7 paginiTim Fields Master Validation Planmanoj262400/2100% (1)
- Validation Master PlanDocument5 paginiValidation Master Planazamyn83% (6)
- Rankine Theory PDFDocument23 paginiRankine Theory PDFHamza NadeemÎncă nu există evaluări
- Guideline On General Principles of Process ValidationDocument15 paginiGuideline On General Principles of Process ValidationRambabu komati - QAÎncă nu există evaluări
- Empower 3 Enterprise or Workgroup Software For ServersDocument52 paginiEmpower 3 Enterprise or Workgroup Software For ServersMohamed Sallam100% (2)
- 014 Quality Unit Roles and ResponsibilitiesDocument35 pagini014 Quality Unit Roles and ResponsibilitiesSIRAJ KP100% (1)
- FDA Approach To AuditingDocument38 paginiFDA Approach To Auditingkjdir100% (2)
- Computer System Validation in The Perspective of T PDFDocument7 paginiComputer System Validation in The Perspective of T PDFFkÎncă nu există evaluări
- Validation Master Plan SummaryDocument25 paginiValidation Master Plan SummaryAtul Sharma100% (2)
- GMP Supplier Assessment QuestionnaireDocument2 paginiGMP Supplier Assessment Questionnairedrs_mdu48Încă nu există evaluări
- An Approach To Process ValidationDocument17 paginiAn Approach To Process ValidationBibek Singh Mahat100% (4)
- Cover LetterDocument7 paginiCover Letterrohan guptaÎncă nu există evaluări
- Computer System ValidationDocument2 paginiComputer System ValidationVishal VakilÎncă nu există evaluări
- 2015 Validation Protocol TemplateDocument8 pagini2015 Validation Protocol TemplateJitesh M Nair100% (1)
- F03qa038-00 VMPDocument24 paginiF03qa038-00 VMPMeet Vermaa100% (1)
- SOP - 0400 - 10 - Design Qualification SOPDocument13 paginiSOP - 0400 - 10 - Design Qualification SOPsandipkumardshahÎncă nu există evaluări
- Cleaning Validation Master Plan PDFDocument9 paginiCleaning Validation Master Plan PDFBREWSKIÎncă nu există evaluări
- Installation Qualification Template IQDocument25 paginiInstallation Qualification Template IQSheila Bersamin TabuconÎncă nu există evaluări
- Star Fleet Ships of The Four Years War, Volume 2Document28 paginiStar Fleet Ships of The Four Years War, Volume 2Rob RobertsÎncă nu există evaluări
- S 264 Validation Spreadsheet ApplicationsDocument14 paginiS 264 Validation Spreadsheet ApplicationsNeoÎncă nu există evaluări
- Basics of Equipment Qualification - Pharma Pathway PDFDocument6 paginiBasics of Equipment Qualification - Pharma Pathway PDFJ VENKATESHÎncă nu există evaluări
- Validation ProtocolDocument63 paginiValidation ProtocolIndústria Petys64% (22)
- Validation Master Plan Annex 15Document29 paginiValidation Master Plan Annex 15spark80988100% (6)
- Computer System Validation Basic Documentation PackageDocument5 paginiComputer System Validation Basic Documentation Packageakaribasappa75% (4)
- HR BestPracticesDocument62 paginiHR BestPracticessaravanans100% (10)
- Guideline For The Transfer of Analytical Test ProceduresDocument5 paginiGuideline For The Transfer of Analytical Test Proceduresjljimenez1969100% (5)
- Installation Qualification For Informatic System ExampleDocument7 paginiInstallation Qualification For Informatic System ExampleCarlos SanchezÎncă nu există evaluări
- THE AND: Fixed Stars Constellations AstrologyDocument258 paginiTHE AND: Fixed Stars Constellations AstrologyВукашин Б Васић100% (1)
- Validation Master Plan TemplateDocument17 paginiValidation Master Plan TemplateNadine100% (4)
- Site Validation Master Plan OverviewDocument34 paginiSite Validation Master Plan OverviewJonatan Dominguez PerezÎncă nu există evaluări
- Process Validation ProtocolDocument10 paginiProcess Validation ProtocolDivya SekarÎncă nu există evaluări
- 6.E Installation Qualification (IQ) : Here You Will Find Answers To The Following QuestionsDocument10 pagini6.E Installation Qualification (IQ) : Here You Will Find Answers To The Following QuestionsFilipÎncă nu există evaluări
- Validation Master Plan A Complete Guide - 2021 EditionDe la EverandValidation Master Plan A Complete Guide - 2021 EditionÎncă nu există evaluări
- 2015 VMP TemplateDocument10 pagini2015 VMP Templatekulbhushan singh100% (2)
- Analytical Procedure Validation Manual 041 SampleDocument3 paginiAnalytical Procedure Validation Manual 041 SampleRambabu komati - QAÎncă nu există evaluări
- SOP For Qualification of Vendors - Pharmaceutical GuidelinesDocument3 paginiSOP For Qualification of Vendors - Pharmaceutical Guidelineskavya nainita100% (1)
- Annual Product Quality Review Data Summary and TrendsDocument1 paginăAnnual Product Quality Review Data Summary and Trendsnasreen anjumÎncă nu există evaluări
- Pharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersDe la EverandPharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersÎncă nu există evaluări
- Design QualificationDocument9 paginiDesign Qualificationtrinath16198050% (2)
- AAPS PharmSciTech Review of Manual Inspection PracticesDocument7 paginiAAPS PharmSciTech Review of Manual Inspection Practicesrobin hasanÎncă nu există evaluări
- Validation Master Plan A Complete Guide - 2020 EditionDe la EverandValidation Master Plan A Complete Guide - 2020 EditionÎncă nu există evaluări
- Exercises in Structural DynamicsDocument13 paginiExercises in Structural DynamicsObinna ObiefuleÎncă nu există evaluări
- Computerized System ValidationDocument14 paginiComputerized System ValidationEka OktawigunaÎncă nu există evaluări
- Iq Oq PQ ChamberDocument17 paginiIq Oq PQ Chamberelias_7760% (5)
- Facility Qualification - Book Published by IVTDocument142 paginiFacility Qualification - Book Published by IVTNitinPrachiJain100% (4)
- OPTIMIZE SITE VALIDATION MASTER PLANDocument32 paginiOPTIMIZE SITE VALIDATION MASTER PLANnophadonÎncă nu există evaluări
- Iq, Oq, PQ, DQDocument17 paginiIq, Oq, PQ, DQsiruslara649167% (3)
- CIQA Validation Master Plan Sample TemplateDocument4 paginiCIQA Validation Master Plan Sample TemplateSatyam Gupta100% (1)
- 4 - Case Study On A Risk-Based Approach To Validation - For ReviewDocument49 pagini4 - Case Study On A Risk-Based Approach To Validation - For Reviewpate malabanan100% (1)
- Validation of HPLC Techniques For Pharmaceutical AnalysisDocument17 paginiValidation of HPLC Techniques For Pharmaceutical AnalysisAhmad Abdullah Najjar100% (14)
- Friability Tester IQ ProtocolDocument25 paginiFriability Tester IQ ProtocolJames Huang75% (4)
- Site Master FileDocument6 paginiSite Master FileRambabu komati - QA100% (3)
- HVAC QualificationDocument36 paginiHVAC QualificationMuhammadAteeq100% (3)
- Analytical Method ValidationDocument86 paginiAnalytical Method ValidationRambabu komati - QA100% (12)
- Facility ValidationDocument12 paginiFacility ValidationGhanta Ranjith Kumar100% (1)
- Laboratory Quality Agreement TamplateDocument10 paginiLaboratory Quality Agreement TamplateMina Maher MikhailÎncă nu există evaluări
- Risk-Based Validation and Requalification of Processes & EquipmentDocument33 paginiRisk-Based Validation and Requalification of Processes & EquipmentfenixseravgonÎncă nu există evaluări
- Validation of Automated Systems & Software - A Practical ApproachDocument24 paginiValidation of Automated Systems & Software - A Practical ApproachLudy YohanaÎncă nu există evaluări
- HPLC ValidationDocument15 paginiHPLC ValidationRambabu komati - QA100% (5)
- Computer System Validation Definition and RequirementsDocument3 paginiComputer System Validation Definition and RequirementsmeongÎncă nu există evaluări
- Validation PolicyDocument3 paginiValidation PolicyneppoanandÎncă nu există evaluări
- Bio-Validation of Steam Sterilization - 758572676Document11 paginiBio-Validation of Steam Sterilization - 758572676venkats_001Încă nu există evaluări
- Method ValidationDocument16 paginiMethod ValidationRambabu komati - QA100% (3)
- 10072-02 RQ Protocol Line-1 Tunnel-001Document34 pagini10072-02 RQ Protocol Line-1 Tunnel-001deepanmb00750% (2)
- SCADA Process Control System User RequirementsDocument65 paginiSCADA Process Control System User Requirementskapernikov50% (2)
- 3850 Mathematics Stage 3 Marking Guide Sample 2Document1 pagină3850 Mathematics Stage 3 Marking Guide Sample 2AshleeGedeliahÎncă nu există evaluări
- Validation Plan TemplateDocument23 paginiValidation Plan Templatedes50% (2)
- Validation Protocol CG TADocument30 paginiValidation Protocol CG TACarlo Duran100% (1)
- The Right To A Balanced and Healthful Ecology by Antonio G.M. La ViñaDocument30 paginiThe Right To A Balanced and Healthful Ecology by Antonio G.M. La Viñaellen joy chanÎncă nu există evaluări
- Froms Guide Lines 150Document5 paginiFroms Guide Lines 150Rambabu komati - QAÎncă nu există evaluări
- Change ManagementDocument28 paginiChange ManagementRambabu komati - QAÎncă nu există evaluări
- 10 Minutes With DR Abdul J KalamDocument11 pagini10 Minutes With DR Abdul J KalamArun Kumar KanaujiaÎncă nu există evaluări
- The Basis Facts of Cleaning ValidationDocument8 paginiThe Basis Facts of Cleaning Validationjljimenez1969100% (1)
- HR Policies 180Document10 paginiHR Policies 180nomi1975100% (2)
- Numbering SystemDocument8 paginiNumbering SystemRambabu komati - QA0% (1)
- eCTD Leaflet EupharDocument2 paginieCTD Leaflet EupharRambabu komati - QAÎncă nu există evaluări
- Guideline On Dossier Requirements For Type - 1A N 1BDocument40 paginiGuideline On Dossier Requirements For Type - 1A N 1BRambabu komati - QA100% (1)
- Esubmissions Requirements New Applications 1Document2 paginiEsubmissions Requirements New Applications 1Rambabu komati - QAÎncă nu există evaluări
- Nees Emea GuidanceDocument31 paginiNees Emea GuidancenkszoneÎncă nu există evaluări
- Europe DMF GuidelinesDocument17 paginiEurope DMF GuidelinesRambabu komati - QA100% (1)
- Col CareDocument5 paginiCol Care829255Încă nu există evaluări
- Top 10 Deficiencies of Dossiers - EDQMDocument4 paginiTop 10 Deficiencies of Dossiers - EDQMRambabu komati - QA100% (4)
- The Ten Principles of GMPDocument3 paginiThe Ten Principles of GMPRambabu komati - QA100% (3)
- Guideline On Plastic Immediate Packaging Material - EMEADocument11 paginiGuideline On Plastic Immediate Packaging Material - EMEAmasthan6yÎncă nu există evaluări
- D80AR Quality GuidanceDocument25 paginiD80AR Quality GuidanceRambabu komati - QAÎncă nu există evaluări
- Ten Commandments of GMPDocument1 paginăTen Commandments of GMPRambabu komati - QA100% (3)
- GMP MeansDocument1 paginăGMP MeansRambabu komati - QAÎncă nu există evaluări
- Shortcuts To SuccessDocument1 paginăShortcuts To SuccessRambabu komati - QAÎncă nu există evaluări
- Exchange 2013 High Availability and Site ResilienceDocument4 paginiExchange 2013 High Availability and Site ResilienceKhodor AkoumÎncă nu există evaluări
- Costco Cover Letter ExamplesDocument6 paginiCostco Cover Letter Examplesxwxxoyvhf100% (1)
- Open Channel Laboratory ExperimentDocument3 paginiOpen Channel Laboratory ExperimentJohn Ceasar PascoÎncă nu există evaluări
- Scope of The StudyDocument13 paginiScope of The StudyKather ShaÎncă nu există evaluări
- Aho - Indexed GrammarsDocument25 paginiAho - Indexed GrammarsgizliiiiÎncă nu există evaluări
- Teaching Goal Examples: Retrieved From Http://sitemaker - Umich.edu/ginapchaney/teaching - Goals - PhilosophyDocument1 paginăTeaching Goal Examples: Retrieved From Http://sitemaker - Umich.edu/ginapchaney/teaching - Goals - PhilosophyFang GohÎncă nu există evaluări
- HemeDocument9 paginiHemeCadenzaÎncă nu există evaluări
- Lethe Berner Keep Warm Drawer BHW 70 2 MANDocument17 paginiLethe Berner Keep Warm Drawer BHW 70 2 MANGolden OdyesseyÎncă nu există evaluări
- ZMMR34Document25 paginiZMMR34Himanshu PrasadÎncă nu există evaluări
- Harvard Negotiation Insight Initiative Program on Negotiation at Harvard Law School Summer Learning Forum The EnneagramDocument1 paginăHarvard Negotiation Insight Initiative Program on Negotiation at Harvard Law School Summer Learning Forum The EnneagramRay GurdÎncă nu există evaluări
- The Learner Understands The Guidelines and Criteria in The Selection and Evaluation of Health InformationDocument3 paginiThe Learner Understands The Guidelines and Criteria in The Selection and Evaluation of Health InformationANACORITA O. SILAGANÎncă nu există evaluări
- SPH Mass ConservationDocument15 paginiSPH Mass ConservationnahkbceÎncă nu există evaluări
- Post Const CalcDocument57 paginiPost Const Calcal mooreÎncă nu există evaluări
- National Territory of the Philippines Defined in <40 CharactersDocument13 paginiNational Territory of the Philippines Defined in <40 CharactersMarianne AngÎncă nu există evaluări
- The Truth Is A Thorny Issue'1 Lesbian Denial in Jackie Kay's TrumpetDocument12 paginiThe Truth Is A Thorny Issue'1 Lesbian Denial in Jackie Kay's TrumpetTahir KhanÎncă nu există evaluări
- Collecting Information & Forecasting DemandDocument34 paginiCollecting Information & Forecasting DemandCinta NastasyaÎncă nu există evaluări
- Refractory Materials For Slagging GasifiersDocument24 paginiRefractory Materials For Slagging GasifiersMian GaoÎncă nu există evaluări
- Koolhaas SpotCheckConversation 1999Document21 paginiKoolhaas SpotCheckConversation 1999Lijani GriesselÎncă nu există evaluări
- 4 Listgarten M. Electron Microscopic Study of The Gingivodental Junction of Man. Am J Anat 1966 119-147 (HWN)Document31 pagini4 Listgarten M. Electron Microscopic Study of The Gingivodental Junction of Man. Am J Anat 1966 119-147 (HWN)Benjamin NgÎncă nu există evaluări
- Raiseplus Weekly Plan English Subject - Sample Only Region 5Document4 paginiRaiseplus Weekly Plan English Subject - Sample Only Region 5Jenelyn Ramos SantiagoÎncă nu există evaluări
- MAM INS 30.4 Tabla Calculo Caida de MaterialesDocument2 paginiMAM INS 30.4 Tabla Calculo Caida de Materialeslourdes Tapia AlfaroÎncă nu există evaluări
- Soukup 2002Document7 paginiSoukup 2002Rodrigo DaliaÎncă nu există evaluări
- Nihilism's Challenge to Meaning and ValuesDocument2 paginiNihilism's Challenge to Meaning and ValuesancorsaÎncă nu există evaluări