S-ar putea să vă placă și
- Solid CatalystsDocument8 paginiSolid CatalystsVISHAL SHARMAÎncă nu există evaluări
- Handouts PDFDocument53 paginiHandouts PDFSandeep ChallaÎncă nu există evaluări
- Micp PDFDocument31 paginiMicp PDFAngela rismaÎncă nu există evaluări
- Knudsen EffusionDocument9 paginiKnudsen EffusionMohammadÎncă nu există evaluări
- L12 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocument25 paginiL12 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiMehul VarshneyÎncă nu există evaluări
- Density Logging FinalDocument8 paginiDensity Logging FinalAvighyan ChakrabortyÎncă nu există evaluări
- L16 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocument25 paginiL16 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiOjasvi MongaÎncă nu există evaluări
- NDX OlsonDocument31 paginiNDX OlsonnaeemdehrajÎncă nu există evaluări
- TP3 - Ceramics: Sintering and Microstructure: Responsable: Abhishek Kumar (MXC 230, Tel: 36888)Document15 paginiTP3 - Ceramics: Sintering and Microstructure: Responsable: Abhishek Kumar (MXC 230, Tel: 36888)neko706Încă nu există evaluări
- Chemical Reaction Engineering-II (2170501) : Semester - VII (CHEM) Chapter Name: Catalysis (Chapter 5)Document11 paginiChemical Reaction Engineering-II (2170501) : Semester - VII (CHEM) Chapter Name: Catalysis (Chapter 5)khushbu100% (2)
- 0705 2894 PDFDocument14 pagini0705 2894 PDFHassanImranÎncă nu există evaluări
- Chapter 9 Thin Film Deposition - IIDocument35 paginiChapter 9 Thin Film Deposition - IIFahmi Alfian SyahÎncă nu există evaluări
- R.-H. Yoon: Minerals EngineeringDocument12 paginiR.-H. Yoon: Minerals EngineeringFrancisco CampbellÎncă nu există evaluări
- L16 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocument25 paginiL16 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiMehul VarshneyÎncă nu există evaluări
- Lecture 5. Neutron LoggingDocument72 paginiLecture 5. Neutron LoggingDimash AbdrakhmanovÎncă nu există evaluări
- Effect of Some Additions On The Sinterability and Magnetic Properties of Barium HexaferriteDocument10 paginiEffect of Some Additions On The Sinterability and Magnetic Properties of Barium HexaferriteMohammedÎncă nu există evaluări
- Ch14iglandkmtonline 101031113719 Phpapp01Document47 paginiCh14iglandkmtonline 101031113719 Phpapp01Ahmed AimanÎncă nu există evaluări
- Blaine's ExperimentDocument8 paginiBlaine's Experimentmunazzil.cscÎncă nu există evaluări
- Keyhole Stability During Laser Welding Part 2Document10 paginiKeyhole Stability During Laser Welding Part 2Jacques OosthuizenÎncă nu există evaluări
- Blast Furnace Aero Dynamics NewDocument36 paginiBlast Furnace Aero Dynamics NewSandeepÎncă nu există evaluări
- Chapter Two Reservoir Properties PorosityDocument35 paginiChapter Two Reservoir Properties Porositymohamed sadekÎncă nu există evaluări
- Dynamic Surface Tension of Micellar Solutions Studied by The Maximum Bubble Pressure Method - ExperimentDocument12 paginiDynamic Surface Tension of Micellar Solutions Studied by The Maximum Bubble Pressure Method - Experimentjvchique100% (1)
- Multiple Saturation QuestionsDocument14 paginiMultiple Saturation QuestionsrestofficalÎncă nu există evaluări
- 03 Catalyst CharacterizationDocument39 pagini03 Catalyst CharacterizationMegan TorresÎncă nu există evaluări
- 5 - Flow Through Packed BedsDocument24 pagini5 - Flow Through Packed BedsاشرفاللساميÎncă nu există evaluări
- Application of The Wheeler-Jonas Equation For The Calculation of Carbon Monolith Breakthrough TimesDocument9 paginiApplication of The Wheeler-Jonas Equation For The Calculation of Carbon Monolith Breakthrough TimeslucashckÎncă nu există evaluări
- Density Log: GS, Sudan Oct. 2005Document21 paginiDensity Log: GS, Sudan Oct. 2005Firdaus Dauz SÎncă nu există evaluări
- CHE 631: Chemical Reaction Engineering: Animangsu GhatakDocument11 paginiCHE 631: Chemical Reaction Engineering: Animangsu GhatakPankaj Kumar SainiÎncă nu există evaluări
- Experimental Testing of Hydrodynamic Collision Model in Fine Particle FlotationDocument6 paginiExperimental Testing of Hydrodynamic Collision Model in Fine Particle Flotationjsotofmet4918Încă nu există evaluări
- Breakup of Gas Bubbles Rising in Molten MetalsDocument6 paginiBreakup of Gas Bubbles Rising in Molten Metalsmaa bloÎncă nu există evaluări
- Shrinking core model for gas-solid reactionsDocument45 paginiShrinking core model for gas-solid reactionsDemas JatiÎncă nu există evaluări
- 4RA34RB3 Lecture Note-8 HPGe DetectorDocument20 pagini4RA34RB3 Lecture Note-8 HPGe DetectorSlide ShareÎncă nu există evaluări
- Intrinsic Structural-Ballistic Interactions in Composite Energetic Materials Part Ii - ModelingDocument15 paginiIntrinsic Structural-Ballistic Interactions in Composite Energetic Materials Part Ii - Modelinggsaucedoz2857Încă nu există evaluări
- Porosity LogDocument15 paginiPorosity Logmts1234Încă nu există evaluări
- Diffusion Limitations in Fischer-Tropsch CatalystsDocument8 paginiDiffusion Limitations in Fischer-Tropsch CatalystsCarolina BermúdezÎncă nu există evaluări
- Chapter 5 Solid and FluidDocument50 paginiChapter 5 Solid and FluidJerome FizzowÎncă nu există evaluări
- Porosity Log: Types and UsesDocument30 paginiPorosity Log: Types and UsesSubh vermaÎncă nu există evaluări
- Hydrogen Gas Absorber Made of Manganese DioxideDocument3 paginiHydrogen Gas Absorber Made of Manganese DioxideAysan JameeÎncă nu există evaluări
- Evidence Daily RoutinesDocument7 paginiEvidence Daily RoutinesJohanna Santiago GuerreroÎncă nu există evaluări
- Dramatic Effect of Trace Oxide on Liquid Metal Rheology MeasurementsDocument29 paginiDramatic Effect of Trace Oxide on Liquid Metal Rheology MeasurementsKalusha Moron CruzÎncă nu există evaluări
- Adsorption and Pore Condensation of Krypton On MesoporousDocument7 paginiAdsorption and Pore Condensation of Krypton On MesoporousSibnath KayalÎncă nu există evaluări
- Mechanisms of Foam Flow in Porous Media: Apparent Viscosity in Smooth CapillariesDocument15 paginiMechanisms of Foam Flow in Porous Media: Apparent Viscosity in Smooth CapillariesfylypeÎncă nu există evaluări
- Antifoam AgentsDocument37 paginiAntifoam AgentsEleonoraÎncă nu există evaluări
- Mass Transfer Equipment DesignDocument68 paginiMass Transfer Equipment DesignFathihah AnuarÎncă nu există evaluări
- Control of Reactive Sputtering ProcessesDocument17 paginiControl of Reactive Sputtering Processeskarin_inakaÎncă nu există evaluări
- Basic Rock and Fluid PropertiesDocument53 paginiBasic Rock and Fluid PropertieskimoÎncă nu există evaluări
- 1990 - Groves e Zhang - A Dilatation Model For The Expansion ofDocument8 pagini1990 - Groves e Zhang - A Dilatation Model For The Expansion ofclanardinoÎncă nu există evaluări
- The Marsh cone: a rheological test or apparatusDocument8 paginiThe Marsh cone: a rheological test or apparatusMostafa Hussein AbdouÎncă nu există evaluări
- Grease InterceptorDocument11 paginiGrease Interceptor185412Încă nu există evaluări
- Formation Evaluation Measurements: ENM237 Reservoir Geology and PetrophysicsDocument67 paginiFormation Evaluation Measurements: ENM237 Reservoir Geology and PetrophysicsMustapha BouregaaÎncă nu există evaluări
- CH-440 NanotechnologyDocument22 paginiCH-440 NanotechnologyAndrew SionÎncă nu există evaluări
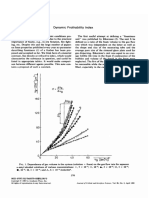
- Dynamic Frothability IndexDocument3 paginiDynamic Frothability IndexjvchiqueÎncă nu există evaluări
- Introduction to Reservoir Petrophysics (By Shoaib and BilalDocument115 paginiIntroduction to Reservoir Petrophysics (By Shoaib and BilalChukwunoso Nwonye100% (5)
- Open Hole Logging Tool: Bulk Density LogDocument12 paginiOpen Hole Logging Tool: Bulk Density LogVenkataramana AvulaÎncă nu există evaluări
- TtipngkkgDocument12 paginiTtipngkkgاسماعيل الرجاميÎncă nu există evaluări
- A Talk On Characteristics of Density Logs in Hydrocarbon IdentificationDocument42 paginiA Talk On Characteristics of Density Logs in Hydrocarbon IdentificationShan ShahzadÎncă nu există evaluări
- Disc Bowl CentrifugeDocument8 paginiDisc Bowl CentrifugeYu HuiÎncă nu există evaluări
- Fundamentals of Fluidized-Bed Chemical Processes: Butterworths Monographs in Chemical EngineeringDe la EverandFundamentals of Fluidized-Bed Chemical Processes: Butterworths Monographs in Chemical EngineeringÎncă nu există evaluări
- Festkörper Probleme VIII: Advances in Solid State PhysicsDe la EverandFestkörper Probleme VIII: Advances in Solid State PhysicsÎncă nu există evaluări
- Grobler Scalable 2015 Dual Fluidised Bed.Document98 paginiGrobler Scalable 2015 Dual Fluidised Bed.Godfrey Eric MuendoÎncă nu există evaluări
- Applied Energy: Gang Xu, Shengwei Huang, Yongping Yang, Ying Wu, Kai Zhang, Cheng XuDocument11 paginiApplied Energy: Gang Xu, Shengwei Huang, Yongping Yang, Ying Wu, Kai Zhang, Cheng XuGodfrey Eric MuendoÎncă nu există evaluări
- Fuel Processing Technology: K. Papadikis, S. Gu, A.V. BridgwaterDocument12 paginiFuel Processing Technology: K. Papadikis, S. Gu, A.V. BridgwaterGodfrey Eric MuendoÎncă nu există evaluări
- Soft Matter: PaperDocument7 paginiSoft Matter: PaperGodfrey Eric MuendoÎncă nu există evaluări
- Introduction To Catalysis Index 2014Document6 paginiIntroduction To Catalysis Index 2014Godfrey Eric MuendoÎncă nu există evaluări
- Integrated Particle-And Reactor-Scale Simulation of Pine Pyrolysis in A Fluidized BedDocument12 paginiIntegrated Particle-And Reactor-Scale Simulation of Pine Pyrolysis in A Fluidized BedGodfrey Eric MuendoÎncă nu există evaluări
- Catalysts: Selective Catalytic Reduction of NODocument4 paginiCatalysts: Selective Catalytic Reduction of NOGodfrey Eric MuendoÎncă nu există evaluări
- Şerbănescu2014 Article KineticAnalysisOfCellulosePyroDocument14 paginiŞerbănescu2014 Article KineticAnalysisOfCellulosePyroGodfrey Eric MuendoÎncă nu există evaluări
- A.V. Bridgewater (1999) An Overview of Fast Pyrolysis of BiomassDocument15 paginiA.V. Bridgewater (1999) An Overview of Fast Pyrolysis of BiomassPieter van der MeerÎncă nu există evaluări
- Soft Matter: PaperDocument7 paginiSoft Matter: PaperGodfrey Eric MuendoÎncă nu există evaluări
- Introduction To Catalysis Cp7Document17 paginiIntroduction To Catalysis Cp7Godfrey Eric MuendoÎncă nu există evaluări
- Introduction To Catalysis Cp6Document37 paginiIntroduction To Catalysis Cp6Godfrey Eric MuendoÎncă nu există evaluări
- Catalysis TodayDocument16 paginiCatalysis TodayGodfrey Eric MuendoÎncă nu există evaluări
- CSC 411 Project Module Reactor Design ParametersDocument2 paginiCSC 411 Project Module Reactor Design ParametersGodfrey Eric MuendoÎncă nu există evaluări
- Green Synthesis of Hydrotalcite From Untreated Mag-Johan LabushcagneDocument13 paginiGreen Synthesis of Hydrotalcite From Untreated Mag-Johan LabushcagneGodfrey Eric MuendoÎncă nu există evaluări
- Catalyst Deactivation Causes and StrategiesDocument26 paginiCatalyst Deactivation Causes and StrategiesGodfrey Eric MuendoÎncă nu există evaluări
- Introduction To Catalysis Cp1Document13 paginiIntroduction To Catalysis Cp1Godfrey Eric MuendoÎncă nu există evaluări
- Introduction To Catalysis Cp5Document13 paginiIntroduction To Catalysis Cp5Godfrey Eric MuendoÎncă nu există evaluări
- Kinetics of Heterogeneous Catalyzed ReactionsDocument49 paginiKinetics of Heterogeneous Catalyzed ReactionsGodfrey Eric MuendoÎncă nu există evaluări
- Catalysts 08 00384 SelectivityDocument12 paginiCatalysts 08 00384 SelectivityGodfrey Eric MuendoÎncă nu există evaluări
- Photocatalytic Removal of Nitrogen Oxides Via TitaDocument6 paginiPhotocatalytic Removal of Nitrogen Oxides Via TitaGodfrey Eric MuendoÎncă nu există evaluări
- Zn-Al Layered Double Hydroxides As Efficient Photocatalysts For Nox AbatementDocument9 paginiZn-Al Layered Double Hydroxides As Efficient Photocatalysts For Nox AbatementGodfrey Eric MuendoÎncă nu există evaluări
- Photodecomposition of NOx on Ag/TiO2 composite catalystsDocument8 paginiPhotodecomposition of NOx on Ag/TiO2 composite catalystsGodfrey Eric MuendoÎncă nu există evaluări
- Catalysts 09 00170 Electron BandDocument18 paginiCatalysts 09 00170 Electron BandGodfrey Eric MuendoÎncă nu există evaluări
- Areviewonphotocatalysisforairtreatment FromcatalystdevelopmenttoreactordesignDocument24 paginiAreviewonphotocatalysisforairtreatment FromcatalystdevelopmenttoreactordesignGodfrey Eric MuendoÎncă nu există evaluări
- 324 11 Zumreoglu Karan AuthorDocument11 pagini324 11 Zumreoglu Karan AuthorGodfrey Eric MuendoÎncă nu există evaluări
- Research Article: Photocatalytic Degradation of No Using Ni-Containing TioDocument8 paginiResearch Article: Photocatalytic Degradation of No Using Ni-Containing TioGodfrey Eric MuendoÎncă nu există evaluări
- 1 s2.0 S0920586118315104 Main - AnnotatedDocument12 pagini1 s2.0 S0920586118315104 Main - AnnotatedGodfrey Eric MuendoÎncă nu există evaluări
- Curvature CorrectionDocument7 paginiCurvature CorrectionE J SÎncă nu există evaluări
- Vibrations and Man's ResponseDocument3 paginiVibrations and Man's ResponsemonoplomoÎncă nu există evaluări
- APSC 100 Module 2 Lab 6 - Chemical EngineeringDocument11 paginiAPSC 100 Module 2 Lab 6 - Chemical EngineeringIan IpÎncă nu există evaluări
- 2k15-ES Lec-5 3D Forces SystemDocument31 pagini2k15-ES Lec-5 3D Forces SystemUsama KhanÎncă nu există evaluări
- Low Energy Antimatter Physics: Marco GiammarchiDocument20 paginiLow Energy Antimatter Physics: Marco GiammarchiLeonardo MonacoÎncă nu există evaluări
- Lab 7 Enzyme KineticsDocument6 paginiLab 7 Enzyme KineticsErikRodriguezÎncă nu există evaluări
- Fluid MCQDocument45 paginiFluid MCQdivyeshÎncă nu există evaluări
- Class X - A - Physics # TEST - 1 - 21.12.2016Document4 paginiClass X - A - Physics # TEST - 1 - 21.12.2016Sankar KumarasamyÎncă nu există evaluări
- Buckling TestDocument11 paginiBuckling Testsharusli100% (1)
- Jurnal Urea Formaldehyde 1Document9 paginiJurnal Urea Formaldehyde 1Afriyanti RosmadianaÎncă nu există evaluări
- CAE DS – High Pressure Die Casting Design Gate and VentingDocument13 paginiCAE DS – High Pressure Die Casting Design Gate and VentingAndrey Polyakov75% (4)
- Chapter 10tifDocument41 paginiChapter 10tifManP130% (1)
- Chapter 20 PDFDocument58 paginiChapter 20 PDFSergio Luis Guerra SánchezÎncă nu există evaluări
- Experiment 108 Transverse Waves - Frequency of Vibration - Online ModifiedDocument6 paginiExperiment 108 Transverse Waves - Frequency of Vibration - Online ModifiedBlank BlankÎncă nu există evaluări
- Air Permeability: CHE1022: Mechanical Operation Lab Expt No: 9 DateDocument4 paginiAir Permeability: CHE1022: Mechanical Operation Lab Expt No: 9 Date;(Încă nu există evaluări
- CHE+3054S+Solid+Fluid+Reactor+Design+ST+Ver+2+ Print-FriendlyDocument31 paginiCHE+3054S+Solid+Fluid+Reactor+Design+ST+Ver+2+ Print-FriendlynmhatityeÎncă nu există evaluări
- Chapter 28 Liquid Chromatography: (D) Reversed-Phase PackingDocument4 paginiChapter 28 Liquid Chromatography: (D) Reversed-Phase PackingElaine P.Încă nu există evaluări
- Ziegler-Nichols Controller Tuning ExampleDocument6 paginiZiegler-Nichols Controller Tuning ExampleChristine AvdikouÎncă nu există evaluări
- Ce2305 Foundation Engineering 2 Marks Questions & Answers 16 Marks QuestionsDocument11 paginiCe2305 Foundation Engineering 2 Marks Questions & Answers 16 Marks Questionsanandk792Încă nu există evaluări
- Class 10 Physics ExplorerDocument30 paginiClass 10 Physics ExplorerRajendra PatelÎncă nu există evaluări
- A2 Chapter 14 OscillationDocument61 paginiA2 Chapter 14 OscillationkwaikunÎncă nu există evaluări
- Collisions in One Dimension LabDocument3 paginiCollisions in One Dimension Labmahmoud2mirza100% (1)
- 3D Numerical Calculation Electric FieldDocument8 pagini3D Numerical Calculation Electric FieldCesar ZamudioÎncă nu există evaluări
- Meru University Homework on Morphic Resonance FieldsDocument4 paginiMeru University Homework on Morphic Resonance FieldsAkshaya Kumar Rath100% (1)
- Single Crystals, Powders and TwinsDocument48 paginiSingle Crystals, Powders and TwinsJabbar AkbarÎncă nu există evaluări
- Units of MeassurementDocument98 paginiUnits of MeassurementRichmond AlcalaÎncă nu există evaluări
- Welding TerminologyDocument5 paginiWelding TerminologyKaruppiah ArunachalamÎncă nu există evaluări
- NMR Manual For Evans MethodDocument3 paginiNMR Manual For Evans MethodTamara Chachi-Baía do SantiagoÎncă nu există evaluări
- Engr: Kamran Soomro MCAT/ECAT-2016Document4 paginiEngr: Kamran Soomro MCAT/ECAT-2016Kamran AliÎncă nu există evaluări
- 1000 Electrical Machines MCQsDocument458 pagini1000 Electrical Machines MCQskibrom atsbha100% (4)