S-ar putea să vă placă și
- 2018 ThermogravimetricAnalysisofPolymersBookChapter PDFDocument30 pagini2018 ThermogravimetricAnalysisofPolymersBookChapter PDFA1234 AJEFÎncă nu există evaluări
- 2017 - Fatigue of Short Fiber Thermoplastic Composites A Review of Recent Experimental Results and AnalysisDocument13 pagini2017 - Fatigue of Short Fiber Thermoplastic Composites A Review of Recent Experimental Results and AnalysisSubramani PichandiÎncă nu există evaluări
- Thermomechanics of Shape Memory Polymer Nanocomposites: Yiping Liu, Ken Gall, Martin L. Dunn, Patrick MccluskeyDocument12 paginiThermomechanics of Shape Memory Polymer Nanocomposites: Yiping Liu, Ken Gall, Martin L. Dunn, Patrick MccluskeyRajendra Prasad DashÎncă nu există evaluări
- Materials Letters: Zhengdao Wang, Weibin Song, Liaoliang Ke, Yuesheng WangDocument3 paginiMaterials Letters: Zhengdao Wang, Weibin Song, Liaoliang Ke, Yuesheng WangKudzie Craig Kelvin MutasaÎncă nu există evaluări
- Shape Memory ShubhamDocument11 paginiShape Memory Shubhamtimroddick24Încă nu există evaluări
- Shape Memory PolymerDocument29 paginiShape Memory PolymerkostarasÎncă nu există evaluări
- Tempera CoupAM Solids&Structures04Document17 paginiTempera CoupAM Solids&Structures04adeodatoarthur2020Încă nu există evaluări
- Boyd 1996Document38 paginiBoyd 1996Avinash KumarÎncă nu există evaluări
- Jrisso Enief2004Document16 paginiJrisso Enief2004Jose RissoÎncă nu există evaluări
- International Journal of Solids and Structures: Young-Ik Yoo, Young-Jin Kim, Dong-Kil Shin, Jung-Ju LeeDocument11 paginiInternational Journal of Solids and Structures: Young-Ik Yoo, Young-Jin Kim, Dong-Kil Shin, Jung-Ju LeeMarité MalachevskyÎncă nu există evaluări
- Residual Stresses in Thermoplastic Composites A ST PDFDocument16 paginiResidual Stresses in Thermoplastic Composites A ST PDFILHAM HUZAINILÎncă nu există evaluări
- Takaki 2013Document12 paginiTakaki 2013fdcarazoÎncă nu există evaluări
- J Jmps 2005 03 009Document27 paginiJ Jmps 2005 03 009mohammad hosein benvidiÎncă nu există evaluări
- Thermomechanics of Shape Memory Polymers: Uniaxial Experiments and Constitutive ModelingDocument35 paginiThermomechanics of Shape Memory Polymers: Uniaxial Experiments and Constitutive ModelingVeeturiVarunÎncă nu există evaluări
- A Nonequilibrium Irreversible Thermodynamic 2007 International Journal of SoDocument26 paginiA Nonequilibrium Irreversible Thermodynamic 2007 International Journal of So姚志豪Încă nu există evaluări
- English 1Document13 paginiEnglish 1amin_127Încă nu există evaluări
- Prediction of Thermal Fatigue Life of A Turbine Nozzle Guide VaneDocument9 paginiPrediction of Thermal Fatigue Life of A Turbine Nozzle Guide VanejswxieÎncă nu există evaluări
- 04 01 19 PDFDocument10 pagini04 01 19 PDFSergiy KulmanÎncă nu există evaluări
- A Constitutive Model For Transformation, Reorientation and Plastic Deformation of Shape Memory AlloysDocument14 paginiA Constitutive Model For Transformation, Reorientation and Plastic Deformation of Shape Memory AlloysMudavath BaburamÎncă nu există evaluări
- Creep ViscoelastDocument12 paginiCreep Viscoelastlrodriguez_892566Încă nu există evaluări
- Bergstrom Bischoff Final 2010Document9 paginiBergstrom Bischoff Final 2010usamaumerÎncă nu există evaluări
- Martensitic Transformations in Shape-Memory Alloys. Successes and Failures of Thermal Analysis and CalorimetryDocument26 paginiMartensitic Transformations in Shape-Memory Alloys. Successes and Failures of Thermal Analysis and CalorimetrySrinivasan NarayananÎncă nu există evaluări
- Modeling and Experimental Verification of Martensitic Transformation Plastic Behavior in Carbon Steel For Quenching ProcessDocument5 paginiModeling and Experimental Verification of Martensitic Transformation Plastic Behavior in Carbon Steel For Quenching ProcessDanish KhanÎncă nu există evaluări
- Propellants Explo Pyrotec - 2022 - Lafourcade - Elastic Anisotropy of 1 3 5 Triamino 2 4 6 Trinitrobenzene As A Function ofDocument12 paginiPropellants Explo Pyrotec - 2022 - Lafourcade - Elastic Anisotropy of 1 3 5 Triamino 2 4 6 Trinitrobenzene As A Function ofasdfasdfasdfÎncă nu există evaluări
- Helm 2001Document12 paginiHelm 2001Avinash KumarÎncă nu există evaluări
- Landau Phase-Field Model For Shape Memory AlloysDocument15 paginiLandau Phase-Field Model For Shape Memory AlloysPrevalisÎncă nu există evaluări
- On Temperature-Related Shift Factors and Master Curves in Viscoelastic Constitutive Models For Thermoset PolymersDocument18 paginiOn Temperature-Related Shift Factors and Master Curves in Viscoelastic Constitutive Models For Thermoset PolymersNam Huu TranÎncă nu există evaluări
- 3 2009 171 Gur ZL p93 103Document12 pagini3 2009 171 Gur ZL p93 103Chiheb BaÎncă nu există evaluări
- Three-Dimensional Model For Solids Undergoing Stress-Induced Phase TransformationsDocument18 paginiThree-Dimensional Model For Solids Undergoing Stress-Induced Phase TransformationsEhab WilsonÎncă nu există evaluări
- 2000 Creep Rupture of A GFRP Composite at Elevated Temperatures - Dutta - Hui - 2000Document9 pagini2000 Creep Rupture of A GFRP Composite at Elevated Temperatures - Dutta - Hui - 2000Rodrigo LameirasÎncă nu există evaluări
- Weld Joints FEADocument26 paginiWeld Joints FEAAlberto darianÎncă nu există evaluări
- An Evaluation of Plastic Flow Stress Models For The Simulation of High-Temperature and High-Strain-Rate Deformation of MetalsDocument43 paginiAn Evaluation of Plastic Flow Stress Models For The Simulation of High-Temperature and High-Strain-Rate Deformation of MetalsMuddu AlaparthiÎncă nu există evaluări
- Rotary Bending Fatigue Analysis of Shape Memory Alloys 2017Document13 paginiRotary Bending Fatigue Analysis of Shape Memory Alloys 2017Israa NizzarÎncă nu există evaluări
- 2016-ASME Turbo16-The Influence of HDB Configuration On Morton Effect - 2016Document10 pagini2016-ASME Turbo16-The Influence of HDB Configuration On Morton Effect - 2016Alex CooperÎncă nu există evaluări
- Time-Temperature Behavior of Carbon - Epoxy Laminates PDFDocument15 paginiTime-Temperature Behavior of Carbon - Epoxy Laminates PDFShabi HadashÎncă nu există evaluări
- Thermo Mechanical Analysis Clean VersionDocument45 paginiThermo Mechanical Analysis Clean VersionHiba MhiriÎncă nu există evaluări
- A Thermodynamic Theory of Short-Term and Creep RuptureDocument6 paginiA Thermodynamic Theory of Short-Term and Creep Ruptureeid elsayedÎncă nu există evaluări
- International Journal of Solids and Structures: Håkan Hallberg, Paul Håkansson, Matti RistinmaaDocument12 paginiInternational Journal of Solids and Structures: Håkan Hallberg, Paul Håkansson, Matti RistinmaaKhouloud GharbiÎncă nu există evaluări
- 2002, B.S. Kim, Numerical Analysis of The Dimensional Stability of Thermoplastic Composites Using A Thermoviscoelastic ApproachDocument15 pagini2002, B.S. Kim, Numerical Analysis of The Dimensional Stability of Thermoplastic Composites Using A Thermoviscoelastic Approachhalil yıldırımÎncă nu există evaluări
- Mechanics of Materials: Kamran A. Khan, Romina Barello, Anastasia H. Muliana, Martin LévesqueDocument18 paginiMechanics of Materials: Kamran A. Khan, Romina Barello, Anastasia H. Muliana, Martin LévesqueLuthfi AdyÎncă nu există evaluări
- Thermo-Magneto-Mechanical Coupling Dynamics of Magnetic Shape Memory AlloysDocument55 paginiThermo-Magneto-Mechanical Coupling Dynamics of Magnetic Shape Memory Alloyszbysław dobiegniewiczÎncă nu există evaluări
- Characterization and Modeling of The Fatigue Behavior of TPUDocument6 paginiCharacterization and Modeling of The Fatigue Behavior of TPUDeepak SharmaÎncă nu există evaluări
- Journal of Thermal StressesDocument30 paginiJournal of Thermal Stressesडॉ. कनिष्क शर्माÎncă nu există evaluări
- TTT CurvesDocument31 paginiTTT CurvesNico PonsÎncă nu există evaluări
- Thermal Stability of Shape Memory Polymers, Polymer Blends, and CompositesDocument31 paginiThermal Stability of Shape Memory Polymers, Polymer Blends, and CompositesVeeturiVarunÎncă nu există evaluări
- 22 Material ModelingDocument10 pagini22 Material ModelingLuis OrtizÎncă nu există evaluări
- SMA ModelingDocument21 paginiSMA ModelingRohit GajbhiyeÎncă nu există evaluări
- Aging McKennaBookChapterdddDocument73 paginiAging McKennaBookChapterdddDinesh SdÎncă nu există evaluări
- 1 s2.0 0749641989900259 MainDocument36 pagini1 s2.0 0749641989900259 Mainxr ChristopherÎncă nu există evaluări
- Rasouli Yazdi1998Document11 paginiRasouli Yazdi1998Antonio Alonso Diaz ArriagaÎncă nu există evaluări
- Lecture 10: Introduction To Theory of Plasticity: Dept. of Mechanical Engg., NIT CalicutDocument10 paginiLecture 10: Introduction To Theory of Plasticity: Dept. of Mechanical Engg., NIT CalicutManoj MallickÎncă nu există evaluări
- PFM MTDocument10 paginiPFM MThemusujithÎncă nu există evaluări
- A Finite Element Model For Shape Memory Alloys Co 2009 International JournalDocument16 paginiA Finite Element Model For Shape Memory Alloys Co 2009 International JournalAvinash KumarÎncă nu există evaluări
- Modulated-Temperature Thermomechanical Analysis: D. M. PriceDocument8 paginiModulated-Temperature Thermomechanical Analysis: D. M. PriceRizky EkoÎncă nu există evaluări
- 2023 08 13T123254.637Document10 pagini2023 08 13T123254.637Mahdi abanÎncă nu există evaluări
- 2013 - Finite Element Simulation On Thermal Fatigue of A Turbine Blade Withthermal Barrier CoatingsDocument10 pagini2013 - Finite Element Simulation On Thermal Fatigue of A Turbine Blade Withthermal Barrier CoatingssumitÎncă nu există evaluări
- Modeling The "Shrink-Wrap Effect" in Polymers and Elastomers, Including The Influence of Very Large Elastic and Inelastic StrainsDocument15 paginiModeling The "Shrink-Wrap Effect" in Polymers and Elastomers, Including The Influence of Very Large Elastic and Inelastic Strainsklomps_jrÎncă nu există evaluări
- 9970 15771 1 SMDocument6 pagini9970 15771 1 SMrxx1218Încă nu există evaluări
- Eyring TheoryDocument5 paginiEyring Theorymariasp95Încă nu există evaluări
- Impact Tensile Behavior Analysis of Twaron Fiber Tows From Fast Fourier TransformDocument9 paginiImpact Tensile Behavior Analysis of Twaron Fiber Tows From Fast Fourier TransformVeeturiVarunÎncă nu există evaluări
- Jin2012 Article EffectsOfLightStabilizerOnTheUDocument7 paginiJin2012 Article EffectsOfLightStabilizerOnTheUVeeturiVarunÎncă nu există evaluări
- Applied SciencesDocument17 paginiApplied SciencesVeeturiVarunÎncă nu există evaluări
- International Journal of Impact Engineering: E. Tapie, V.P.W. Shim, Y.B. GuoDocument12 paginiInternational Journal of Impact Engineering: E. Tapie, V.P.W. Shim, Y.B. GuoVeeturiVarunÎncă nu există evaluări
- Ballistic Performance of Unidirectional Glass Fiber Laminated Composite Plate Under Normal and Oblique ImpactDocument8 paginiBallistic Performance of Unidirectional Glass Fiber Laminated Composite Plate Under Normal and Oblique ImpactVeeturiVarunÎncă nu există evaluări
- Comparison of Ballistic Impact Behavior of Carbon Fiber/Epoxy Composite and Steel Metal StructuresDocument10 paginiComparison of Ballistic Impact Behavior of Carbon Fiber/Epoxy Composite and Steel Metal StructuresVeeturiVarunÎncă nu există evaluări
- Zhu 1998Document19 paginiZhu 1998VeeturiVarunÎncă nu există evaluări
- Polymers 12 00711 PDFDocument13 paginiPolymers 12 00711 PDFVeeturiVarunÎncă nu există evaluări
- Crystals 09 00389 PDFDocument10 paginiCrystals 09 00389 PDFVeeturiVarunÎncă nu există evaluări
- Phadnis 2013 J. Phys. Conf. Ser. 451 012019Document9 paginiPhadnis 2013 J. Phys. Conf. Ser. 451 012019VeeturiVarunÎncă nu există evaluări
- Thermomechanics of Shape Memory Polymers: Uniaxial Experiments and Constitutive ModelingDocument35 paginiThermomechanics of Shape Memory Polymers: Uniaxial Experiments and Constitutive ModelingVeeturiVarunÎncă nu există evaluări
- Prediction of Impact Damage in Composite Plates: J.P. Hou, N. Petrinic, C. Ruiz, S.R. HallettDocument9 paginiPrediction of Impact Damage in Composite Plates: J.P. Hou, N. Petrinic, C. Ruiz, S.R. HallettVeeturiVarunÎncă nu există evaluări
- Direct Correlation Between Modulus and The Crystalline Structure in Isotactic PolypropyleneDocument13 paginiDirect Correlation Between Modulus and The Crystalline Structure in Isotactic PolypropyleneVeeturiVarunÎncă nu există evaluări
- Otymer Communications: Ly Induced Changes in Kevlar" Fi Urfaee EvidencedDocument3 paginiOtymer Communications: Ly Induced Changes in Kevlar" Fi Urfaee EvidencedVeeturiVarunÎncă nu există evaluări
- Annurev Ms 25 080195 001455 PDFDocument31 paginiAnnurev Ms 25 080195 001455 PDFVeeturiVarunÎncă nu există evaluări
- 1995 TC QuasicrystalsDocument5 pagini1995 TC QuasicrystalsVeeturiVarunÎncă nu există evaluări
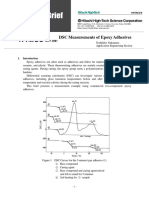
- DSC Measurements of Epoxy Adhesives: Temp. (°C)Document4 paginiDSC Measurements of Epoxy Adhesives: Temp. (°C)VeeturiVarunÎncă nu există evaluări
- Effect of Accelerated Ageing Conditions On The Degradation Process of Dyneema (R) Polyethylene CompositesDocument7 paginiEffect of Accelerated Ageing Conditions On The Degradation Process of Dyneema (R) Polyethylene CompositesVeeturiVarunÎncă nu există evaluări
- Thank You!: Here Is Your ConfirmationDocument2 paginiThank You!: Here Is Your ConfirmationVeeturiVarunÎncă nu există evaluări
- Xie2009 Facile Tailioring of Thermal Transiton Temperatures of Epoxy Shape Memory PolymersDocument5 paginiXie2009 Facile Tailioring of Thermal Transiton Temperatures of Epoxy Shape Memory PolymersVeeturiVarunÎncă nu există evaluări
- Composites: Part B: Yanju Liu, Di Song, Chunxia Wu, Jinsong LengDocument7 paginiComposites: Part B: Yanju Liu, Di Song, Chunxia Wu, Jinsong LengVeeturiVarunÎncă nu există evaluări
- Thermal Stability of Shape Memory Polymers, Polymer Blends, and CompositesDocument31 paginiThermal Stability of Shape Memory Polymers, Polymer Blends, and CompositesVeeturiVarunÎncă nu există evaluări
- Composites Science and Technology: Peng Wang, Shengnan Geng, Tianhuai DingDocument3 paginiComposites Science and Technology: Peng Wang, Shengnan Geng, Tianhuai DingVeeturiVarunÎncă nu există evaluări
- Pedestrian Volume Studies: A Case Study in The City of GothenburgDocument80 paginiPedestrian Volume Studies: A Case Study in The City of GothenburgPaula A. FigueroaÎncă nu există evaluări
- Warrick 26m Control de Nivel AuxiliarDocument3 paginiWarrick 26m Control de Nivel AuxiliarCarlos WayÎncă nu există evaluări
- AQA GCSE Chemistry: 2.1.5 Metallic BondingDocument1 paginăAQA GCSE Chemistry: 2.1.5 Metallic BondingZehmilÎncă nu există evaluări
- GPS 2Document6 paginiGPS 2Arnold Alexander GarzonÎncă nu există evaluări
- Newton First Law of MotionDocument6 paginiNewton First Law of MotionKin ChristineÎncă nu există evaluări
- ECODRIVE - ошибки и предупрежденияDocument6 paginiECODRIVE - ошибки и предупрежденияАндрей ПетряхинÎncă nu există evaluări
- 9011 VW Rebar Strainmeter (E)Document2 pagini9011 VW Rebar Strainmeter (E)JasonÎncă nu există evaluări
- Ebook Remote Server ManagementDocument10 paginiEbook Remote Server ManagementPiruletoÎncă nu există evaluări
- UNIT - 3: Fast-Fourier-Transform (FFT) Algorithms: Dr. Manjunatha. PDocument100 paginiUNIT - 3: Fast-Fourier-Transform (FFT) Algorithms: Dr. Manjunatha. PMVRajeshMaliyeckalÎncă nu există evaluări
- Ec1102n - Mimi Nur Nabila Binti Alias - 2019293128 - Final ExamDocument17 paginiEc1102n - Mimi Nur Nabila Binti Alias - 2019293128 - Final ExammimiÎncă nu există evaluări
- VentilationDocument3 paginiVentilationasdasaÎncă nu există evaluări
- CAT 2020 QUANT Previous Year QuestionsDocument49 paginiCAT 2020 QUANT Previous Year QuestionsApoorva SharmaÎncă nu există evaluări
- 3D ShapesDocument5 pagini3D Shapesdeez000Încă nu există evaluări
- NCERT Solutions For Class 12 Physics Chapter 12 AtomsDocument14 paginiNCERT Solutions For Class 12 Physics Chapter 12 AtomsKritika MishraÎncă nu există evaluări
- Chemical Bonding and Molecular Structure: 2.1. Fundamental Concepts of Chemical BondsDocument47 paginiChemical Bonding and Molecular Structure: 2.1. Fundamental Concepts of Chemical BondsNguyễn Quốc HưngÎncă nu există evaluări
- Re - (Repeater-Builder) Midland Vehicular Repeater InfoDocument3 paginiRe - (Repeater-Builder) Midland Vehicular Repeater InfobbarinkÎncă nu există evaluări
- Mark Scheme (Results) October 2020: Pearson Edexcel International Advanced Level in Core Mathematics C34 (WMA02) Paper 01Document29 paginiMark Scheme (Results) October 2020: Pearson Edexcel International Advanced Level in Core Mathematics C34 (WMA02) Paper 01nonÎncă nu există evaluări
- Project, Program and Portfolio SelectionDocument40 paginiProject, Program and Portfolio Selectionsaif ur rehman shahid hussain (aviator)Încă nu există evaluări
- Siegel57Nonparametric PDFDocument8 paginiSiegel57Nonparametric PDFTarisna AryantiÎncă nu există evaluări
- Module 3 Q2 Gen Chem IIDocument10 paginiModule 3 Q2 Gen Chem IIMengieÎncă nu există evaluări
- Zanussi Zou 342 User ManualDocument12 paginiZanussi Zou 342 User Manualadatok2Încă nu există evaluări
- Datasheet en 20170526Document9 paginiDatasheet en 20170526LODELBARRIO RDÎncă nu există evaluări
- The Effecto of Using Deductive ApproachDocument15 paginiThe Effecto of Using Deductive ApproachCarmela EstradaÎncă nu există evaluări
- Unit QuestionsDocument155 paginiUnit QuestionsSanya KhanÎncă nu există evaluări
- Circular Motion 1 QPDocument8 paginiCircular Motion 1 QPNovi Juniati PitrianaÎncă nu există evaluări
- When Bad Things Happen To Good MissilesDocument9 paginiWhen Bad Things Happen To Good Missilesmykingboody2156Încă nu există evaluări
- Spectral FFT Max Ms PDocument17 paginiSpectral FFT Max Ms Phockey66patÎncă nu există evaluări
- Stephanie Florescu - MPM2D Summary SheetDocument2 paginiStephanie Florescu - MPM2D Summary Sheetstephanie.florescuÎncă nu există evaluări
- Lutron / Trane Bacnet Integration: QuantumDocument7 paginiLutron / Trane Bacnet Integration: QuantumthomasÎncă nu există evaluări
- Jig & Fixture DesignDocument22 paginiJig & Fixture Designmagi017Încă nu există evaluări