S-ar putea să vă placă și
- Computer System Validation in The Perspective of T PDFDocument7 paginiComputer System Validation in The Perspective of T PDFFkÎncă nu există evaluări
- PQE - General Principles of CVSDocument30 paginiPQE - General Principles of CVSDra CupuÎncă nu există evaluări
- Retrospective Validation of A CDS Feb 99 PDFDocument9 paginiRetrospective Validation of A CDS Feb 99 PDFImran AsgharÎncă nu există evaluări
- Data Integrity and Compliance: A Primer for Medical Product ManufacturersDe la EverandData Integrity and Compliance: A Primer for Medical Product ManufacturersÎncă nu există evaluări
- S 285 Risk Qualification InfstructureDocument10 paginiS 285 Risk Qualification InfstructureMohamed SallamÎncă nu există evaluări
- 4 - Case Study On A Risk-Based Approach To Validation - For ReviewDocument49 pagini4 - Case Study On A Risk-Based Approach To Validation - For Reviewpate malabanan100% (1)
- GAMP 5 A Risk Based Approach To A Risk BDocument29 paginiGAMP 5 A Risk Based Approach To A Risk BLia LiawatiÎncă nu există evaluări
- A Study of Computerized System ValidationDocument12 paginiA Study of Computerized System ValidationTenzin Tashi100% (1)
- Understanding CSV User RequirementsDocument27 paginiUnderstanding CSV User RequirementsSivaramakrishna Markandeya Gupta0% (1)
- Data Integrity ChecklistDocument12 paginiData Integrity ChecklistprakashÎncă nu există evaluări
- Computer System ValidationDocument2 paginiComputer System ValidationVishal VakilÎncă nu există evaluări
- SOP For Computer System Validation in Pharmaceutical IndustryDocument8 paginiSOP For Computer System Validation in Pharmaceutical IndustryDeepakÎncă nu există evaluări
- Computer System ValidationDocument10 paginiComputer System ValidationRudra RahmanÎncă nu există evaluări
- ECA Computerised System Validation GAMP 5 ApproachDocument6 paginiECA Computerised System Validation GAMP 5 ApproachHanan NoussaÎncă nu există evaluări
- 1 - Introduction To Computerized Systems Validation - For ReviewDocument41 pagini1 - Introduction To Computerized Systems Validation - For Reviewpate malabananÎncă nu există evaluări
- CSV SopDocument1 paginăCSV SopjeetÎncă nu există evaluări
- Computer System Validation Risk Assessment ToolDocument3 paginiComputer System Validation Risk Assessment Toolcpkakope100% (1)
- Empower 3 21 CFR Part 11 Compliance Assessment RevA November 2019Document14 paginiEmpower 3 21 CFR Part 11 Compliance Assessment RevA November 2019Nur AcarÎncă nu există evaluări
- Periodic ReviewDocument6 paginiPeriodic ReviewJorge GutierrezÎncă nu există evaluări
- Validation of COTS Using Single Life Cycle Approach - RD McDowallDocument15 paginiValidation of COTS Using Single Life Cycle Approach - RD McDowallNitin KashyapÎncă nu există evaluări
- Computer System Validation (CSV) : Comparisons Between (GMP VS CGMP, GLP VS GCP, 21 CFR PART 11 VS EU 11)Document4 paginiComputer System Validation (CSV) : Comparisons Between (GMP VS CGMP, GLP VS GCP, 21 CFR PART 11 VS EU 11)T 1Încă nu există evaluări
- Guidance For Industry CSV 201312Document16 paginiGuidance For Industry CSV 201312Maruf Rassel100% (1)
- Part 11 System Compliance Assessment Checklist: Fda Compliance Digest Published by EnkapDocument3 paginiPart 11 System Compliance Assessment Checklist: Fda Compliance Digest Published by EnkapSairam KishoreÎncă nu există evaluări
- VAL 135 Risk Assessment For Computer Validation Systems Sample - SandraDocument3 paginiVAL 135 Risk Assessment For Computer Validation Systems Sample - SandraSandra Silva100% (1)
- Gamp StandardsDocument6 paginiGamp Standardszaman_rafiqÎncă nu există evaluări
- Sequence of Activities Status Timelines/Key Challenges Computer System Validation For SAP S/4 HANADocument5 paginiSequence of Activities Status Timelines/Key Challenges Computer System Validation For SAP S/4 HANASourav Ghosh DastidarÎncă nu există evaluări
- MS 21 CFR Part 11 RequirementsDocument8 paginiMS 21 CFR Part 11 RequirementsmagtrolÎncă nu există evaluări
- V Model & Validation Process-In The Pharmaceutical Industry - FDA PerspectiveDocument8 paginiV Model & Validation Process-In The Pharmaceutical Industry - FDA Perspectivewholidi3281Încă nu există evaluări
- COMPUTER SYSTEM VALIDATION MASTER PLAN - Pharmaceutical Guidance - RecognizedDocument57 paginiCOMPUTER SYSTEM VALIDATION MASTER PLAN - Pharmaceutical Guidance - Recognizedcpkakope67% (3)
- IQ Consortium Data Integrity Risk Assessment Tool: Click Here To Enter TextDocument13 paginiIQ Consortium Data Integrity Risk Assessment Tool: Click Here To Enter Textjai soni100% (1)
- CSV Risks Requirements Tests and TraceabilityDocument44 paginiCSV Risks Requirements Tests and TraceabilitymonsepackÎncă nu există evaluări
- QRM For Computer SystemsDocument51 paginiQRM For Computer SystemsbaluchakpÎncă nu există evaluări
- Testing Computers Systems For FDA Mhra Compliance Computer Systems Val - ebooKOIDDocument136 paginiTesting Computers Systems For FDA Mhra Compliance Computer Systems Val - ebooKOIDquangterumo100% (10)
- Basic Concepts of CSVDocument23 paginiBasic Concepts of CSVAhmed Samir100% (1)
- Gamp5 For Basic Training PDFDocument47 paginiGamp5 For Basic Training PDFVimlesh Kumar PandeyÎncă nu există evaluări
- Computer System Validation Definition and RequirementsDocument3 paginiComputer System Validation Definition and RequirementsmeongÎncă nu există evaluări
- Data Integrity Related Observation PDFDocument27 paginiData Integrity Related Observation PDFHemant SankhalaÎncă nu există evaluări
- SOP of URS 1657011596Document5 paginiSOP of URS 1657011596Huỳnh Ngọc SángÎncă nu există evaluări
- 21 CFR Part 11 ComplianceDocument2 pagini21 CFR Part 11 Compliancepham hoang quanÎncă nu există evaluări
- CSV Good Documentation and Test Practices For GXPDocument16 paginiCSV Good Documentation and Test Practices For GXPDivyendu DasÎncă nu există evaluări
- Computersystemsvalidation BlogspotDocument7 paginiComputersystemsvalidation BlogspotNitin KashyapÎncă nu există evaluări
- Eres Annex 11 Eu GMP SiemensDocument30 paginiEres Annex 11 Eu GMP SiemenshuykhiemÎncă nu există evaluări
- GMPAnnex 11 ChecklistDocument11 paginiGMPAnnex 11 Checklistosamakamel1Încă nu există evaluări
- Overview of Validation Documents and ProjectsDocument5 paginiOverview of Validation Documents and ProjectsMD Fahad MiajiÎncă nu există evaluări
- Electronic Batch Recording: Kevin Walls Senior Solutions ConsultantDocument68 paginiElectronic Batch Recording: Kevin Walls Senior Solutions ConsultantFaress RabiÎncă nu există evaluări
- Azure FDA 21 CFR Part 11 Qualification Guideline PDFDocument79 paginiAzure FDA 21 CFR Part 11 Qualification Guideline PDFGopal KandasÎncă nu există evaluări
- Computer System ValidationDocument2 paginiComputer System Validationanon_544896262Încă nu există evaluări
- FDA Expectation On Software ValidationDocument40 paginiFDA Expectation On Software ValidationHong HuangÎncă nu există evaluări
- Risk Assessment Example For CSV - EDMSDocument2 paginiRisk Assessment Example For CSV - EDMSvenki_bee100% (2)
- FORM-000249181 DI Equipment Software Pre-Assessment QuestionnaireDocument3 paginiFORM-000249181 DI Equipment Software Pre-Assessment QuestionnaireSebastian LopezÎncă nu există evaluări
- Computer Sytems ValidationDocument92 paginiComputer Sytems Validationiabureid7460100% (16)
- MHRA GXP Data Integrity ConsultationDocument14 paginiMHRA GXP Data Integrity ConsultationBhagyesh Kulakrni100% (1)
- Gamp 5Document5 paginiGamp 5vignan50Încă nu există evaluări
- Computer System Validation - Definition and Requirements - MustRead PDFDocument3 paginiComputer System Validation - Definition and Requirements - MustRead PDFtraining validÎncă nu există evaluări
- Computer Validation StandardsDocument21 paginiComputer Validation Standardssaidvaret100% (1)
- Computer System Validation Basic Documentation PackageDocument5 paginiComputer System Validation Basic Documentation Packageakaribasappa75% (4)
- CSV Full Document PDFDocument39 paginiCSV Full Document PDFAbhijeet100% (1)
- PDFDocument115 paginiPDFCarlos Gene QuirozÎncă nu există evaluări
- OECD Guidelines On Human Biobanks and Genetic Research DatabasesDocument53 paginiOECD Guidelines On Human Biobanks and Genetic Research Databasesttugce29Încă nu există evaluări
- OECD Guidelines On Human Biobanks and Genetic Research DatabasesDocument53 paginiOECD Guidelines On Human Biobanks and Genetic Research Databasesttugce29Încă nu există evaluări
- Avrupa Komi̇syonuDocument72 paginiAvrupa Komi̇syonuttugce29Încă nu există evaluări
- Corrective Actions Background of Terms: EUROLAB "Cook Book" - Doc No. 16Document7 paginiCorrective Actions Background of Terms: EUROLAB "Cook Book" - Doc No. 16ttugce29Încă nu există evaluări
- Ilac Bi̇yobanka BroşürüDocument4 paginiIlac Bi̇yobanka Broşürüttugce29100% (1)
- Biobank Quality Management in The BBMRI - Be NetworkDocument9 paginiBiobank Quality Management in The BBMRI - Be Networkttugce29Încă nu există evaluări
- Bilgisayarlı SistemlerDocument14 paginiBilgisayarlı Sistemlerttugce29Încă nu există evaluări
- Uncertainty From Sampling - NORDTEST Report - TR604Document82 paginiUncertainty From Sampling - NORDTEST Report - TR604Anonymous LEVNDh4Încă nu există evaluări
- UKAS - Technical Bulletin - ISO 17034 - 2016Document2 paginiUKAS - Technical Bulletin - ISO 17034 - 2016ttugce29Încă nu există evaluări
- Eurolab Handbook Iso Iec 17025 2017Document32 paginiEurolab Handbook Iso Iec 17025 2017Ali ZafarÎncă nu există evaluări
- Ea 4 21 Inf Rev00 March 18Document11 paginiEa 4 21 Inf Rev00 March 18ttugce29Încă nu există evaluări
- Uncertainty From Sampling - NORDTEST Report - TR604Document82 paginiUncertainty From Sampling - NORDTEST Report - TR604Anonymous LEVNDh4Încă nu există evaluări
- Remco 2009 PDFDocument12 paginiRemco 2009 PDFttugce29Încă nu există evaluări
- Bilgisayarlı Sistemler SözlüğüDocument72 paginiBilgisayarlı Sistemler Sözlüğüttugce29Încă nu există evaluări
- ISO - IEC 17025 - 2017 - First ImpressionsDocument2 paginiISO - IEC 17025 - 2017 - First Impressionsttugce29Încă nu există evaluări
- Pharh!Aceutica Acta Helvetiae: Daniel Friedli A, Wolfgang Kappeler B, Susanne Zimmermann BDocument6 paginiPharh!Aceutica Acta Helvetiae: Daniel Friedli A, Wolfgang Kappeler B, Susanne Zimmermann Bttugce29Încă nu există evaluări
- Eurolab Handbook Iso Iec 17025 2017Document32 paginiEurolab Handbook Iso Iec 17025 2017Ali ZafarÎncă nu există evaluări
- Data Integrity Checklist PDFDocument5 paginiData Integrity Checklist PDFttugce29100% (1)
- Türki̇yede Referans Malzeme Üreti̇mi̇ PDFDocument4 paginiTürki̇yede Referans Malzeme Üreti̇mi̇ PDFttugce29Încă nu există evaluări
- Iso Proc 33Document12 paginiIso Proc 33ttugce29Încă nu există evaluări
- Iso CascoDocument11 paginiIso Cascottugce29Încă nu există evaluări
- Guia de Seleccion U y Uso de MRDocument10 paginiGuia de Seleccion U y Uso de MREdgar Nina VelasquezÎncă nu există evaluări
- China 568Document54 paginiChina 568ttugce29Încă nu există evaluări
- 1-S2.0-S0950423001000262-Main EoDocument18 pagini1-S2.0-S0950423001000262-Main Eottugce29Încă nu există evaluări
- IUPAC Single-Lab Validation v6 DRAFTDocument30 paginiIUPAC Single-Lab Validation v6 DRAFTttugce29Încă nu există evaluări
- 2011 Annual Report On Financial Assistance For EnlargementDocument203 pagini2011 Annual Report On Financial Assistance For Enlargementttugce29Încă nu există evaluări
- Japan 779Document43 paginiJapan 779ttugce29Încă nu există evaluări
- 1 s2.0 S0376738805001638 MainDocument9 pagini1 s2.0 S0376738805001638 Mainttugce29Încă nu există evaluări
- Service Manual: Multifunction Electrical Tester CalibratorDocument106 paginiService Manual: Multifunction Electrical Tester CalibratorJuan Carlos Ferrer OrtizÎncă nu există evaluări
- Guideline On Smacna Through Penetration Fire StoppingDocument48 paginiGuideline On Smacna Through Penetration Fire Stoppingwguindy70Încă nu există evaluări
- Updoc - Tips Dictionar Foraj e RDocument37 paginiUpdoc - Tips Dictionar Foraj e RDaniela Dandea100% (1)
- ASOTDocument4 paginiASOTemperors_nestÎncă nu există evaluări
- SGT PDFDocument383 paginiSGT PDFDushyanthkumar DasariÎncă nu există evaluări
- Bisleri Water Industry: Project ReportDocument53 paginiBisleri Water Industry: Project ReportJohn CarterÎncă nu există evaluări
- "Next Friend" and "Guardian Ad Litem" - Difference BetweenDocument1 pagină"Next Friend" and "Guardian Ad Litem" - Difference BetweenTeh Hong Xhe100% (2)
- Castle 1-3K E ManualDocument26 paginiCastle 1-3K E ManualShami MudunkotuwaÎncă nu există evaluări
- Behavior Specific Praise Statements HandoutDocument3 paginiBehavior Specific Praise Statements HandoutDaniel BernalÎncă nu există evaluări
- Heteropolyacids FurfuralacetoneDocument12 paginiHeteropolyacids FurfuralacetonecligcodiÎncă nu există evaluări
- Narrative Report On Weekly Accomplishments: Department of EducationDocument2 paginiNarrative Report On Weekly Accomplishments: Department of Educationisha mariano100% (1)
- Phardose Lab Prep 19 30Document4 paginiPhardose Lab Prep 19 30POMPEYO BARROGAÎncă nu există evaluări
- ISO 45001:2018 & OHSAS 18001:2007 Clause-Wise Comparison MatrixDocument3 paginiISO 45001:2018 & OHSAS 18001:2007 Clause-Wise Comparison MatrixvenkatesanÎncă nu există evaluări
- Royal British College IncDocument5 paginiRoyal British College IncLester MojadoÎncă nu există evaluări
- Entitlement Cure SampleDocument34 paginiEntitlement Cure SampleZondervan100% (1)
- ResumeDocument3 paginiResumejohn DaqueÎncă nu există evaluări
- Offender TypologiesDocument8 paginiOffender TypologiesSahil AnsariÎncă nu există evaluări
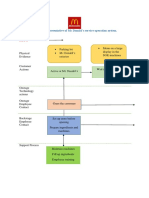
- Blueprint Huynh My Ky Duyen 2022 McDonald'sDocument2 paginiBlueprint Huynh My Ky Duyen 2022 McDonald'sHuỳnh Mỹ Kỳ DuyênÎncă nu există evaluări
- University of Puerto Rico at PonceDocument16 paginiUniversity of Puerto Rico at Ponceapi-583167359Încă nu există evaluări
- Soil Biotechnology (SBT) - Brochure of Life LinkDocument2 paginiSoil Biotechnology (SBT) - Brochure of Life Linkiyer_lakshmananÎncă nu există evaluări
- 2019 06 28 PDFDocument47 pagini2019 06 28 PDFTes BabasaÎncă nu există evaluări
- Intro To Psychological AssessmentDocument7 paginiIntro To Psychological AssessmentKian La100% (1)
- Mil STD 792fDocument13 paginiMil STD 792fdoradoanÎncă nu există evaluări
- Penilaian Akhir TahunDocument4 paginiPenilaian Akhir TahunRestu Suci UtamiÎncă nu există evaluări
- AFMAN91-201 NewDocument458 paginiAFMAN91-201 NewbombtechÎncă nu există evaluări
- Sav4747 PDFDocument49 paginiSav4747 PDFAndres Antonio Moreno CastroÎncă nu există evaluări
- DyslexiaDocument19 paginiDyslexiaKeren HapkhÎncă nu există evaluări
- Chemical Reaction Engineering-II - R2015 - 10-04-2018Document2 paginiChemical Reaction Engineering-II - R2015 - 10-04-201818135A0806 MAKKUVA BHAVYAÎncă nu există evaluări
- Congenital Flexural Deformity in CalfDocument6 paginiCongenital Flexural Deformity in CalfBibek SutradharÎncă nu există evaluări
- 3 Activities For Adults To Practice Modeling SELDocument10 pagini3 Activities For Adults To Practice Modeling SELDavid Garcia PerezÎncă nu există evaluări