S-ar putea să vă placă și
- Fiziopatologie An III - MG - LP12 - RO-1Document8 paginiFiziopatologie An III - MG - LP12 - RO-1yubiiÎncă nu există evaluări
- Curs 8 Fiziopatologie ProteicDocument14 paginiCurs 8 Fiziopatologie ProteicCiprian RataÎncă nu există evaluări
- Biochimie Medicala Curs 2Document31 paginiBiochimie Medicala Curs 2Andreea PetricaÎncă nu există evaluări
- Anemia Macrocitara GATADocument16 paginiAnemia Macrocitara GATAEma ȚurcașÎncă nu există evaluări
- Explorarea Metabolismului ProtidicDocument6 paginiExplorarea Metabolismului ProtidicRusu Stefan100% (1)
- Anemii MegaloblasticeDocument9 paginiAnemii MegaloblasticeElena LupuÎncă nu există evaluări
- Curs 12 Metabolismul ProteicDocument12 paginiCurs 12 Metabolismul ProteicCiotlos AlexandruÎncă nu există evaluări
- SângeleDocument31 paginiSângeleAhmad AlhassanÎncă nu există evaluări
- EritrocitDocument11 paginiEritrocitded_om3nÎncă nu există evaluări
- LP 5 FiziopatologieDocument5 paginiLP 5 FiziopatologiecmvioÎncă nu există evaluări
- ProteineDocument9 paginiProteinemlinaballerinaÎncă nu există evaluări
- Maladii Metabolice UmaneDocument5 paginiMaladii Metabolice Umaneunafantasia92% (12)
- Bolile Inascute de Metabolism IDocument137 paginiBolile Inascute de Metabolism IsilviuburueanaÎncă nu există evaluări
- Anemii 2 - Anemii MacrocitareDocument4 paginiAnemii 2 - Anemii MacrocitareAndrei AngelescuÎncă nu există evaluări
- Curs 1Document55 paginiCurs 1CristinaÎncă nu există evaluări
- Proteine Totale, Electroforeza, Proteine UrinareDocument3 paginiProteine Totale, Electroforeza, Proteine UrinareCristina CrissÎncă nu există evaluări
- Anemia MegaloblasticaDocument4 paginiAnemia MegaloblasticaStroe IonelaÎncă nu există evaluări
- Anemia BiermerDocument25 paginiAnemia BiermerIoana Barcari100% (3)
- 9.proteine Plasmatice-An II - 2020Document65 pagini9.proteine Plasmatice-An II - 2020Larisa StanÎncă nu există evaluări
- Proteine - ScurtDocument5 paginiProteine - ScurtMaria Alexandra GrigoreÎncă nu există evaluări
- Metabolismul ProteicDocument3 paginiMetabolismul ProteicalivinaioanaÎncă nu există evaluări
- Fiziologie - Curs 2 Volumul SanguinDocument6 paginiFiziologie - Curs 2 Volumul Sanguinionut_lupul100% (2)
- Ingrijirea Pacientului Cu Sindrom MalabsorbtieDocument16 paginiIngrijirea Pacientului Cu Sindrom MalabsorbtieNicoleta AmarițeiÎncă nu există evaluări
- Fiziopatologie Curs 5Document4 paginiFiziopatologie Curs 5RoxanaCîlţeaÎncă nu există evaluări
- LP 6 Biochimie Anul I MedicinaDocument6 paginiLP 6 Biochimie Anul I MedicinaAlexandra StoianÎncă nu există evaluări
- Curs 4 Metab - ProtDocument26 paginiCurs 4 Metab - ProtRusu FilofteiaÎncă nu există evaluări
- Cursuri NefrologieDocument156 paginiCursuri NefrologieAlex Cristian Ionut100% (1)
- 8.fiziopatologia Metabolismului ProteicDocument15 pagini8.fiziopatologia Metabolismului ProteicAdÎncă nu există evaluări
- K Anemii Prin Alterarea Sintezei de ADN (MACROcitareDocument26 paginiK Anemii Prin Alterarea Sintezei de ADN (MACROcitareCornelia IlieÎncă nu există evaluări
- Anemia MegaloblasticaDocument4 paginiAnemia Megaloblasticaazygos2008100% (1)
- Explorarea În Laborator Metabolismului Proteic-64119Document61 paginiExplorarea În Laborator Metabolismului Proteic-64119SabinaVascanÎncă nu există evaluări
- ANEMII MegaloblasticeDocument16 paginiANEMII MegaloblasticeUngureanu AndreiÎncă nu există evaluări
- Metabolismul ProteinelorDocument14 paginiMetabolismul ProteinelorcheciudianaÎncă nu există evaluări
- Gastro (Sindromul de Malabsorbtie (SM) )Document6 paginiGastro (Sindromul de Malabsorbtie (SM) )yaser100% (1)
- Hemoliza FinalDocument7 paginiHemoliza FinalizabelutzyÎncă nu există evaluări
- MORFOPAT SPECIALA - TELEMAN 2cap.9Document31 paginiMORFOPAT SPECIALA - TELEMAN 2cap.9Mada BocanceaÎncă nu există evaluări
- Sindromul de MalabsorbtieDocument6 paginiSindromul de MalabsorbtieDaya DavidÎncă nu există evaluări
- MaladiiDocument8 paginiMaladiiAndreea Stefan100% (1)
- LP 7 2020 Fiziopatologie METABOLISM PROTEICDocument20 paginiLP 7 2020 Fiziopatologie METABOLISM PROTEICMihaela-Teodora OanceaÎncă nu există evaluări
- Patologia Asociata SarciniiDocument56 paginiPatologia Asociata SarciniiNelly-Georgiana Mănăilă100% (1)
- Anemia MegaloblasticăDocument5 paginiAnemia MegaloblasticăCiuciu CristinaÎncă nu există evaluări
- Anemia BiermerDocument25 paginiAnemia BiermerAndreea CimpoiÎncă nu există evaluări
- 2 DigestiaDocument15 pagini2 DigestiaAndreea MihaelaÎncă nu există evaluări
- Erori Inascute de Metabolism Released By-MedtorrenDocument58 paginiErori Inascute de Metabolism Released By-MedtorrenVictor JosuÎncă nu există evaluări
- Curs 03. Fiziopatologia Anemiilor, Poliglobuliilor Si Fiziopatologia HemostazeiDocument16 paginiCurs 03. Fiziopatologia Anemiilor, Poliglobuliilor Si Fiziopatologia HemostazeiOlimpiu TiagoÎncă nu există evaluări
- Metabolism ProtidicDocument8 paginiMetabolism ProtidicMyhay CretzuÎncă nu există evaluări
- Insuficienta Hepatica - ScurtDocument7 paginiInsuficienta Hepatica - ScurtKingGeorgeVIIÎncă nu există evaluări
- Eritrocit CAMDocument14 paginiEritrocit CAMAnca AnkÎncă nu există evaluări
- Biochimie FicatDocument29 paginiBiochimie FicatBogdan DreadÎncă nu există evaluări
- Curs 1 Gastroenterologie Ciroza HepaticaDocument35 paginiCurs 1 Gastroenterologie Ciroza HepaticaAndr Crs100% (1)
- Curs 1 Proteine Si Markeri TuDocument37 paginiCurs 1 Proteine Si Markeri TuAndreea AronÎncă nu există evaluări
- Managementul Integrat Al Pacientilor Cu Come Hepatice - StupelimanDocument32 paginiManagementul Integrat Al Pacientilor Cu Come Hepatice - Stupelimanba123asemÎncă nu există evaluări
- Aplazia Medulara2020Document65 paginiAplazia Medulara2020Emilia GuranÎncă nu există evaluări
- Hemato 3 (Anemiile-I II)Document13 paginiHemato 3 (Anemiile-I II)Canal InchisÎncă nu există evaluări
- Eritropoieza Si CoagulareaDocument6 paginiEritropoieza Si Coagulareacristina_zaharia865440Încă nu există evaluări
- Anemia MegaloblasticaDocument29 paginiAnemia MegaloblasticaLuciana MioaraÎncă nu există evaluări
- Dieta pentru zile senine II: De la durere la echilibru spiritual, psihologic, nutritivDe la EverandDieta pentru zile senine II: De la durere la echilibru spiritual, psihologic, nutritivEvaluare: 5 din 5 stele5/5 (1)
- Superintestinul: Un plan de patru săptămâni pentru reprogramarea microbiomului, refacerea sănătății și pierderea în greutateDe la EverandSuperintestinul: Un plan de patru săptămâni pentru reprogramarea microbiomului, refacerea sănătății și pierderea în greutateÎncă nu există evaluări
- Refractia Subiectiva - Recunoasterea OchelkarilorDocument14 paginiRefractia Subiectiva - Recunoasterea Ochelkarilorsufaru ciprianÎncă nu există evaluări
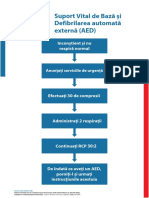
- Poster BLS AutomatedExternal AlgorithmDocument1 paginăPoster BLS AutomatedExternal AlgorithmGuliga NiculinaÎncă nu există evaluări
- Subiecte Stagiu OFTALMODocument27 paginiSubiecte Stagiu OFTALMOsufaru ciprianÎncă nu există evaluări
- Oftalmo Subiecte 1Document44 paginiOftalmo Subiecte 1sufaru ciprianÎncă nu există evaluări
- Curs 7 TraumaDocument52 paginiCurs 7 Traumascrib113Încă nu există evaluări
- Poster BLS AlgorithmDocument1 paginăPoster BLS AlgorithmGuliga NiculinaÎncă nu există evaluări
- Curs 1-2021Document61 paginiCurs 1-2021sufaru ciprianÎncă nu există evaluări
- Diabet Subiecte An VDocument52 paginiDiabet Subiecte An Vsufaru ciprianÎncă nu există evaluări
- Poster SpecCircs Emergency Treatment of Hyperkalaemia AlgorithmDocument1 paginăPoster SpecCircs Emergency Treatment of Hyperkalaemia AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster SpecCircs Drowning AlgorithmDocument1 paginăPoster SpecCircs Drowning AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster SpecCircs Anaphylaxis Treatment AlgorithmDocument1 paginăPoster SpecCircs Anaphylaxis Treatment Algorithmsufaru ciprianÎncă nu există evaluări
- Curs 4-2018Document71 paginiCurs 4-2018Rotaru TeofanaÎncă nu există evaluări
- Poster PALS AlgorithmDocument1 paginăPoster PALS AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster SpecCircs Traumatic Cardiac Arrest AlgorithmDocument1 paginăPoster SpecCircs Traumatic Cardiac Arrest AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster Babies NLS AlgorithmDocument1 paginăPoster Babies NLS AlgorithmGuliga NiculinaÎncă nu există evaluări
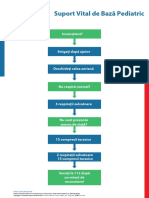
- Poster PAEDS BLS AlgorithmDocument1 paginăPoster PAEDS BLS AlgorithmGuliga NiculinaÎncă nu există evaluări
- Curs 2-2011Document55 paginiCurs 2-2011Sayuridark5Încă nu există evaluări
- Poster SpecCircs Avalanche Accident AlgorithmDocument1 paginăPoster SpecCircs Avalanche Accident AlgorithmGuliga NiculinaÎncă nu există evaluări
- Algoritm Bradicardie PDFDocument1 paginăAlgoritm Bradicardie PDFGuliga NiculinaÎncă nu există evaluări
- Curs 3 2018Document57 paginiCurs 3 2018sufaru ciprianÎncă nu există evaluări
- Poster ALS IHCAT Algorithm Ro PDFDocument1 paginăPoster ALS IHCAT Algorithm Ro PDFGuliga NiculinaÎncă nu există evaluări
- M UDocument62 paginiM UȘtefan ApetriaÎncă nu există evaluări
- Poster ALS Algorithm Ro PDFDocument1 paginăPoster ALS Algorithm Ro PDFGuliga NiculinaÎncă nu există evaluări
- Algoritm TahicardieDocument1 paginăAlgoritm Tahicardiemikaaa000Încă nu există evaluări
- Poster SpecCircs Anaphylaxis Treatment AlgorithmDocument1 paginăPoster SpecCircs Anaphylaxis Treatment Algorithmsufaru ciprianÎncă nu există evaluări
- Poster Babies NLS AlgorithmDocument1 paginăPoster Babies NLS AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster PALS AlgorithmDocument1 paginăPoster PALS AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster SpecCircs Avalanche Accident AlgorithmDocument1 paginăPoster SpecCircs Avalanche Accident AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster PAEDS BLS AlgorithmDocument1 paginăPoster PAEDS BLS AlgorithmGuliga NiculinaÎncă nu există evaluări
- Poster SpecCircs Drowning AlgorithmDocument1 paginăPoster SpecCircs Drowning AlgorithmGuliga NiculinaÎncă nu există evaluări