S-ar putea să vă placă și
- h2020 Hi Oa Data MGT enDocument12 paginih2020 Hi Oa Data MGT enHost MomÎncă nu există evaluări
- Deloitte - DG Report For An Educational Institution PDFDocument172 paginiDeloitte - DG Report For An Educational Institution PDFSumeet AgrawalÎncă nu există evaluări
- Master Governance PlanDocument21 paginiMaster Governance PlanAmgad YassinÎncă nu există evaluări
- Security of Personal Data: The Cnil'S Guides - 2018 EditionDocument24 paginiSecurity of Personal Data: The Cnil'S Guides - 2018 Editionhp2in1i515Încă nu există evaluări
- Guidelines For Data De-Identification or Anonymization - EDUCAUSEDocument33 paginiGuidelines For Data De-Identification or Anonymization - EDUCAUSEHost MomÎncă nu există evaluări
- A Guide To Data GovernanceDocument26 paginiA Guide To Data GovernancesebasgjÎncă nu există evaluări
- Fhir Bulk Data API v10Document42 paginiFhir Bulk Data API v10Host MomÎncă nu există evaluări
- Body of Knowledge: Data AssetsDocument3 paginiBody of Knowledge: Data AssetsHost MomÎncă nu există evaluări
- Body of Knowledge: Governance AssetsDocument7 paginiBody of Knowledge: Governance AssetsHost MomÎncă nu există evaluări
- Data Classification Guide v1.1Document7 paginiData Classification Guide v1.1Host MomÎncă nu există evaluări
- Step 8 - Data Sharing AgreementsDocument2 paginiStep 8 - Data Sharing AgreementsHost MomÎncă nu există evaluări
- Data Protection and Confidentiality Policy 1.0Document23 paginiData Protection and Confidentiality Policy 1.0Host MomÎncă nu există evaluări
- ComplianceForge Hierarchical Cybersecurity Governance FrameworkDocument5 paginiComplianceForge Hierarchical Cybersecurity Governance FrameworkKPÎncă nu există evaluări
- Collibra Staffing Skills MatrixDocument6 paginiCollibra Staffing Skills MatrixAhsan FarooquiÎncă nu există evaluări
- Data Classification Workflow For Data StewardsDocument2 paginiData Classification Workflow For Data StewardsHost MomÎncă nu există evaluări
- Collibra Catalog FactsheetDocument2 paginiCollibra Catalog FactsheetHost MomÎncă nu există evaluări
- Body of Knowledge: Approaches To Reference Data ManagementDocument5 paginiBody of Knowledge: Approaches To Reference Data ManagementHost MomÎncă nu există evaluări
- Body of Knowledge: Approval Process Walk ThroughDocument16 paginiBody of Knowledge: Approval Process Walk ThroughHost MomÎncă nu există evaluări
- Body of Knowledge: Workflow DefinitionsDocument5 paginiBody of Knowledge: Workflow DefinitionsHost MomÎncă nu există evaluări
- Hipaa Gap Analysis 13Document50 paginiHipaa Gap Analysis 13Host MomÎncă nu există evaluări
- Body of Knowledge: OMDP Step 8 - Identify Communities and DomainsDocument7 paginiBody of Knowledge: OMDP Step 8 - Identify Communities and DomainsHost MomÎncă nu există evaluări
- Body of Knowledge: Importing Reference Data From Excel: IsO Country Codes and SubdivisionsDocument12 paginiBody of Knowledge: Importing Reference Data From Excel: IsO Country Codes and SubdivisionsHost MomÎncă nu există evaluări
- HITRUST Policies - Third-Party Assurance ProcedureDocument9 paginiHITRUST Policies - Third-Party Assurance ProcedureHost MomÎncă nu există evaluări
- Vaccine Effectiveness Against SARS-CoV-2 Infection With The Omicron or DeltaDocument6 paginiVaccine Effectiveness Against SARS-CoV-2 Infection With The Omicron or DeltaHost MomÎncă nu există evaluări
- New Vestibu GDPR-01 ComplianceAssessmentDocument2 paginiNew Vestibu GDPR-01 ComplianceAssessmentHost MomÎncă nu există evaluări
- Cnil Ip3 Data Muses and Borders of Creative Arts EngDocument16 paginiCnil Ip3 Data Muses and Borders of Creative Arts EngHost MomÎncă nu există evaluări
- HIPAA-Questionnaire Gap Analysis GridDocument49 paginiHIPAA-Questionnaire Gap Analysis GridHost MomÎncă nu există evaluări
- Strategic Plan: by Your Side To Build A Trusted Digital SocietyDocument8 paginiStrategic Plan: by Your Side To Build A Trusted Digital SocietyHost MomÎncă nu există evaluări
- Cnil Ip Report 06 Shaping Choices in The Digital WorldDocument50 paginiCnil Ip Report 06 Shaping Choices in The Digital WorldSpit FireÎncă nu există evaluări
- New Vestibu GDPR-02 PersonalDataManagementDocument2 paginiNew Vestibu GDPR-02 PersonalDataManagementHost MomÎncă nu există evaluări
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (74)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- D8045-16 Standard Test Method For Acid Number of Crude Oils and Petroleum Products by CatalyticDocument6 paginiD8045-16 Standard Test Method For Acid Number of Crude Oils and Petroleum Products by CatalytichishamÎncă nu există evaluări
- CD Compre Exam 2 Key AnswerDocument6 paginiCD Compre Exam 2 Key AnswerGrace LazaroÎncă nu există evaluări
- Human Nutritional RequirementsDocument3 paginiHuman Nutritional RequirementsAgnesMagadiaÎncă nu există evaluări
- Material Safety Data Sheet Roto-Inject FluidDocument5 paginiMaterial Safety Data Sheet Roto-Inject FluidQuintana JoseÎncă nu există evaluări
- Final Workshop Report On Value Addition and AgroprocessingDocument31 paginiFinal Workshop Report On Value Addition and AgroprocessingBett K. BernardÎncă nu există evaluări
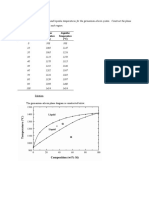
- Composition Solidus Temperature Liquidus Temperature: (WT% Si) (°C) (°C)Document7 paginiComposition Solidus Temperature Liquidus Temperature: (WT% Si) (°C) (°C)Muhammad Ibkar YusranÎncă nu există evaluări
- English 1 Reading (CVC)Document27 paginiEnglish 1 Reading (CVC)Angelica ArcangelÎncă nu există evaluări
- V. Aa. Gram A/S: DescriptionDocument2 paginiV. Aa. Gram A/S: Descriptioncontango O&GÎncă nu există evaluări
- Exp 4 Centrifugal CompressorDocument11 paginiExp 4 Centrifugal CompressorFaris HamirÎncă nu există evaluări
- AE Board Review Part3 - MSWord2003 Long Paper 2011Document12 paginiAE Board Review Part3 - MSWord2003 Long Paper 2011Jan14Încă nu există evaluări
- Dialysis and ElectrodialysisDocument32 paginiDialysis and ElectrodialysisJuan CarvajalÎncă nu există evaluări
- Book Summary - I Hear You Summary Michael Sorensen - Read in 7 MinutesDocument10 paginiBook Summary - I Hear You Summary Michael Sorensen - Read in 7 MinutesNogüey JoseÎncă nu există evaluări
- Complications of Diabetes MellitusDocument46 paginiComplications of Diabetes MellitusAbhijith MenonÎncă nu există evaluări
- 06 - Flexible Operation of Thermal Power Plants - OEM Perspective and Experiences PDFDocument22 pagini06 - Flexible Operation of Thermal Power Plants - OEM Perspective and Experiences PDFRavishankarÎncă nu există evaluări
- Types of ProcurementDocument7 paginiTypes of ProcurementrahulÎncă nu există evaluări
- NWO Plans Exposed by Insider in 1969Document36 paginiNWO Plans Exposed by Insider in 1969Stig Dragholm100% (3)
- Tutorials 2016Document54 paginiTutorials 2016Mankush Jain100% (1)
- Job Satisfaction and Professional Ethics Practices in Public SectorDocument13 paginiJob Satisfaction and Professional Ethics Practices in Public SectorMuhammad NafeesÎncă nu există evaluări
- 1erTareaMicroscopíasLópez Marmolejo Clere MishellDocument4 pagini1erTareaMicroscopíasLópez Marmolejo Clere Mishellclere02marmolejoÎncă nu există evaluări
- MPCA Response 5.29.20Document2 paginiMPCA Response 5.29.20Duluth News TribuneÎncă nu există evaluări
- Megapower: Electrosurgical GeneratorDocument45 paginiMegapower: Electrosurgical GeneratorAnibal Alfaro VillatoroÎncă nu există evaluări
- DemolitionDocument25 paginiDemolitionusler4u100% (1)
- DBM MonetizationDocument2 paginiDBM MonetizationrsdiamzÎncă nu există evaluări
- Genius 7 On Bill Acceptor ManualDocument10 paginiGenius 7 On Bill Acceptor ManualJose Maria PerezÎncă nu există evaluări
- Soal Uh English XDocument1 paginăSoal Uh English XhenniherawatiÎncă nu există evaluări
- Lesson 3.3 Inside An AtomDocument42 paginiLesson 3.3 Inside An AtomReign CallosÎncă nu există evaluări
- Challenges in The Functional Diagnosis of Thyroid Nodules Before Surgery For TSH-producing Pituitary AdenomaDocument5 paginiChallenges in The Functional Diagnosis of Thyroid Nodules Before Surgery For TSH-producing Pituitary AdenomaAthul IgnatiusÎncă nu există evaluări
- A211 Reading Set A QuestionDocument12 paginiA211 Reading Set A Questiontasya zakariaÎncă nu există evaluări
- Polycystic Ovary Syndrome (PCOS) - Symptoms, Causes, and TreatmentDocument19 paginiPolycystic Ovary Syndrome (PCOS) - Symptoms, Causes, and TreatmentAkshay HarekarÎncă nu există evaluări
- Equipment Available From SJ Turbine Inc.: LM Package Parts LM Package Parts High VoltageDocument59 paginiEquipment Available From SJ Turbine Inc.: LM Package Parts LM Package Parts High VoltageAnibal QuezadaÎncă nu există evaluări