S-ar putea să vă placă și
- Informe N°01 Practico de FarmacologíaDocument24 paginiInforme N°01 Practico de FarmacologíaElida Delgado Vasquez100% (1)
- ClonidinaDocument7 paginiClonidinaMerlina VazquezÎncă nu există evaluări
- Aulton - Farmacia La Ciencia Del Diseño de Formas FarmacéuticasDocument686 paginiAulton - Farmacia La Ciencia Del Diseño de Formas FarmacéuticasGloria Miramontes100% (4)
- Course Exercise - Autoevaluación Previa Al Curso de SVAP - SpanishDocument3 paginiCourse Exercise - Autoevaluación Previa Al Curso de SVAP - SpanishPriscila MartìnezÎncă nu există evaluări
- Principios Farmacologia PediatricaDocument153 paginiPrincipios Farmacologia PediatricaJulio César Mendoza100% (1)
- Intoxicacion BenzodiacepinasDocument26 paginiIntoxicacion BenzodiacepinasSharon Angélica Vargas QuiscaÎncă nu există evaluări
- Vías de AdministraciónDocument30 paginiVías de AdministraciónツImSonnyÎncă nu există evaluări
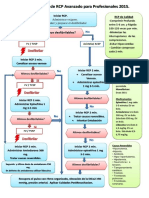
- Algoritmos RCP AvanzadoDocument1 paginăAlgoritmos RCP AvanzadoMaynor Duarte100% (4)
- Spanish Patient Brochure BotoxDocument50 paginiSpanish Patient Brochure BotoxEnderlaser Centro de Cirugía Plástica y Estética0% (1)
- Agonistas ColinérgicosDocument10 paginiAgonistas ColinérgicosManuel MirandaÎncă nu există evaluări
- Influencia Del Entorno Donde Se HabitaDocument7 paginiInfluencia Del Entorno Donde Se HabitaMaria SanchezÎncă nu există evaluări
- Esquizofrenia Desde La Perspectiva Del PacienteDocument2 paginiEsquizofrenia Desde La Perspectiva Del PacienteMaria SanchezÎncă nu există evaluări
- Del Soporte de Autonomia y La Motivacion Docente PDFDocument8 paginiDel Soporte de Autonomia y La Motivacion Docente PDFMaria SanchezÎncă nu există evaluări
- Estudio Del Estado Nutricional de La Poblacion InfantilDocument7 paginiEstudio Del Estado Nutricional de La Poblacion InfantilMaria SanchezÎncă nu există evaluări
- Vigilancia epidemiológica: definición, objetivos y nivelesDocument2 paginiVigilancia epidemiológica: definición, objetivos y nivelesMaria SanchezÎncă nu există evaluări
- Depresion y Obesidad InfantilDocument5 paginiDepresion y Obesidad InfantilMaria SanchezÎncă nu există evaluări
- Cuando La Falta de Inteligencia EmocionalDocument7 paginiCuando La Falta de Inteligencia EmocionalMaria SanchezÎncă nu există evaluări
- Del Soporte de Autonomia y La Motivacion Docente PDFDocument8 paginiDel Soporte de Autonomia y La Motivacion Docente PDFMaria SanchezÎncă nu există evaluări
- Influenza A (H1N1): concepto, agente causal, epidemiología y prevenciónDocument13 paginiInfluenza A (H1N1): concepto, agente causal, epidemiología y prevenciónMaria SanchezÎncă nu există evaluări
- Ejercicios para BrazosDocument10 paginiEjercicios para BrazosMaria SanchezÎncă nu există evaluări
- Enfermedades Respiratorias. ExposicionDocument9 paginiEnfermedades Respiratorias. ExposicionMaria SanchezÎncă nu există evaluări
- Almeja Amarilla InterpretaciónDocument1 paginăAlmeja Amarilla InterpretaciónMaria SanchezÎncă nu există evaluări
- Espinoza Rodriguez Ninive EdiliaDocument82 paginiEspinoza Rodriguez Ninive EdiliaMaria SanchezÎncă nu există evaluări
- Ficha de dexmedetomidina para enfermeríaDocument1 paginăFicha de dexmedetomidina para enfermeríaArturo MartinezÎncă nu există evaluări
- BPD en Establecimientos FarmaceuticosDocument36 paginiBPD en Establecimientos FarmaceuticosRosa Pilar Mamani HilasacaÎncă nu există evaluări
- Prospecto Sulfadim JarabeDocument1 paginăProspecto Sulfadim JarabeValeria PalmieriÎncă nu există evaluări
- Central de Mezclas Farmacéutica: Fcv. Productos Hospitalarios Proceso: ProduccionDocument3 paginiCentral de Mezclas Farmacéutica: Fcv. Productos Hospitalarios Proceso: ProduccionSergio Vergel MantillaÎncă nu există evaluări
- Proyecto AMU FinalDocument10 paginiProyecto AMU FinalMilena del Cisne Santorum ChalacanÎncă nu există evaluări
- Semana 1-2 HistoriaDocument24 paginiSemana 1-2 HistoriaJoubert Diaz MarinÎncă nu există evaluări
- Tipos de Hipotensores OcularesDocument2 paginiTipos de Hipotensores OcularesElfer LópezÎncă nu există evaluări
- Ficha Técnica AnestesicosDocument9 paginiFicha Técnica AnestesicosMaria jose Burbano GonzalezÎncă nu există evaluări
- TMDVDocument29 paginiTMDVValeska ZamoraÎncă nu există evaluări
- PSL Meloxicam 75mg IfavetDocument1 paginăPSL Meloxicam 75mg IfavetLaura SánchezÎncă nu există evaluări
- Consumo de Sildenafil en JovenesDocument2 paginiConsumo de Sildenafil en Jovenesdiseneo6238100% (2)
- Fisiología - Resultados Práctica 1 Vias de AdministracionDocument4 paginiFisiología - Resultados Práctica 1 Vias de AdministracionIvanÎncă nu există evaluări
- Investigacion Bajo Sobre Anesteisa.Document14 paginiInvestigacion Bajo Sobre Anesteisa.danielÎncă nu există evaluări
- NEUROAXIALDocument40 paginiNEUROAXIALNicolás DuránÎncă nu există evaluări
- MetronidazolDocument3 paginiMetronidazolMaria Gabriela RamirezÎncă nu există evaluări
- PALS GUIAS 2010 DR - MuruaDocument35 paginiPALS GUIAS 2010 DR - MuruaJulián Velásquez PÎncă nu există evaluări
- Codigos Color Jeringas PDFDocument1 paginăCodigos Color Jeringas PDFGrupo de Anestesiólogos CUPMÎncă nu există evaluări
- 1 Trabajo Botero y LaguadoDocument14 pagini1 Trabajo Botero y Laguadolitza liliana cabrera cuencaÎncă nu există evaluări
- Inhibidores Selectivos de La COXDocument6 paginiInhibidores Selectivos de La COXCarlos Francisco Mancheno MoncayoÎncă nu există evaluări
- Biofarmacia FarmacocineticaDocument32 paginiBiofarmacia FarmacocineticaSaul Antonio Montoya SerranoÎncă nu există evaluări