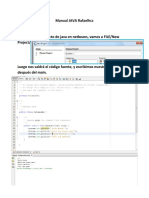
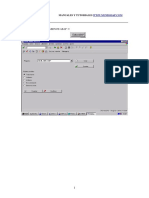
S-ar putea să vă placă și
- Algoritmo de LevenshteinDocument6 paginiAlgoritmo de LevenshteinDemian HiddenÎncă nu există evaluări
- Variable CompleDocument211 paginiVariable CompleMariaÎncă nu există evaluări
- Formato IEEEDocument3 paginiFormato IEEEAdri RMÎncă nu există evaluări
- Ejercicios de Teoría ElectromagneticaDocument36 paginiEjercicios de Teoría ElectromagneticaJohny CampañaÎncă nu există evaluări
- Cuestionario 2Document9 paginiCuestionario 2AntonioLinkEzeroÎncă nu există evaluări
- Resumen Mecánica Clásica I 2020B EPN Aider - Jhon Chiliquinga & Kevin CárdenasDocument110 paginiResumen Mecánica Clásica I 2020B EPN Aider - Jhon Chiliquinga & Kevin CárdenasKAGGHGÎncă nu există evaluări
- Apuntes Sistemas DistribuidosDocument71 paginiApuntes Sistemas DistribuidostoniskaiÎncă nu există evaluări
- Regiones Simple y Multiplemente Conexas - PresentacionDocument21 paginiRegiones Simple y Multiplemente Conexas - Presentacionmarkus_x86Încă nu există evaluări
- Tarea 1 SDC 2015Document22 paginiTarea 1 SDC 2015Salvador Gallardo100% (1)
- Sumadorrestador - Contador 0 99Document24 paginiSumadorrestador - Contador 0 99Jose Ignacio Fajardo AguirreÎncă nu există evaluări
- Formato IEEE Proyecto FinalDocument4 paginiFormato IEEE Proyecto FinaljmdelacruzvÎncă nu există evaluări
- GuiaETS LenguajeDeProgramacionDocument11 paginiGuiaETS LenguajeDeProgramacionexcelencia5Încă nu există evaluări
- ETS Variable ComplejaDocument4 paginiETS Variable Complejaorigen123Încă nu există evaluări
- Dispos 1Document8 paginiDispos 1ArianÎncă nu există evaluări
- Solución de Sistemas de Ecuaciones 4x4 Por DeterminantesDocument8 paginiSolución de Sistemas de Ecuaciones 4x4 Por DeterminantesJose ChochabotÎncă nu există evaluări
- Tarea Nº6 Sistemas Lineales Dinámicos PDFDocument26 paginiTarea Nº6 Sistemas Lineales Dinámicos PDFCamilo Ignacio Arriagada JaraÎncă nu există evaluări
- 03 - Quine McCluskey y KarnaughDocument47 pagini03 - Quine McCluskey y Karnaughalex_cumbalÎncă nu există evaluări
- PWM Usando Dos NE555Document20 paginiPWM Usando Dos NE555Miguel Ángel Flores TicayÎncă nu există evaluări
- Electronica Alta FrecuenciaDocument43 paginiElectronica Alta FrecuenciaEdwin JamiÎncă nu există evaluări
- Analisis Grafico2Document9 paginiAnalisis Grafico2robefiÎncă nu există evaluări
- Codigo TrellisDocument15 paginiCodigo TrellisSILVERACÎncă nu există evaluări
- Clase Práctica Sobre Combinación Lineal y Espacio GeneradoDocument1 paginăClase Práctica Sobre Combinación Lineal y Espacio GeneradoHector TineoÎncă nu există evaluări
- Solucionario de Circuitos Eléctricos en Estado Estable y Transiente Tomo II ESPOCHDocument104 paginiSolucionario de Circuitos Eléctricos en Estado Estable y Transiente Tomo II ESPOCHKAGGHGÎncă nu există evaluări
- Tipos de Datos Clases PDFDocument24 paginiTipos de Datos Clases PDFEva VenturaÎncă nu există evaluări
- Guía de Laboratorio VectoresDocument6 paginiGuía de Laboratorio Vectorestarzan702100% (1)
- Guia Ets 15 2-1Document18 paginiGuia Ets 15 2-1Tavo MalosoÎncă nu există evaluări
- Fortran 90Document29 paginiFortran 90Glenn Robert Revolledo VilchezÎncă nu există evaluări
- Ley de SchmitDocument3 paginiLey de SchmitRocio Alejandra Marin GattoniÎncă nu există evaluări
- Práctica 3Document4 paginiPráctica 3EMÎncă nu există evaluări
- Teoria Electromegnetica Juan Carlos GranadaDocument323 paginiTeoria Electromegnetica Juan Carlos GranadaErika ErikaÎncă nu există evaluări
- 1B Sesión Teoría Intensivo 2021Document41 pagini1B Sesión Teoría Intensivo 2021Manuel Arevalo VillanuevaÎncă nu există evaluări
- Efecto MariposaDocument2 paginiEfecto MariposaManuel Romero100% (1)
- Teorema de GreenDocument23 paginiTeorema de GreenRoonyÎncă nu există evaluări
- Ejercicios 4 y 5Document6 paginiEjercicios 4 y 5Laura Valentina Gaitan GamezÎncă nu există evaluări
- 1° PC Ecuaciones DiferencialesDocument15 pagini1° PC Ecuaciones DiferencialesErnesto Pinedo CastilloÎncă nu există evaluări
- Diseño Digital Multiplicador Con Sumas SucesivasDocument8 paginiDiseño Digital Multiplicador Con Sumas SucesivasLuis HerediaÎncă nu există evaluări
- Laboratorio N°01Document6 paginiLaboratorio N°01Jackeline Pariona Gomez100% (1)
- Electronicos PDFDocument283 paginiElectronicos PDFPatricio JarrínÎncă nu există evaluări
- Apuntes de Física Matemática - Antonio Cañada Villar - Universidad de Granada PDFDocument65 paginiApuntes de Física Matemática - Antonio Cañada Villar - Universidad de Granada PDFAlex HCÎncă nu există evaluări
- Deberes Propuestos Cal-Vect Tres Parciales2018Document20 paginiDeberes Propuestos Cal-Vect Tres Parciales2018josecueva1618Încă nu există evaluări
- Taller 2 PDFDocument30 paginiTaller 2 PDFAnyi Acosta CortésÎncă nu există evaluări
- PIDDocument9 paginiPIDMaria Monica Rodriguez BeltranÎncă nu există evaluări
- Convolucion GraficaDocument13 paginiConvolucion Graficalijoce5586% (7)
- Proyecto Tetris Lenguaje de ProgramacionDocument3 paginiProyecto Tetris Lenguaje de Programacionjesús danielÎncă nu există evaluări
- Pérdidas Debido Al Dieléctrico en Una Guía de Onda RectangularDocument4 paginiPérdidas Debido Al Dieléctrico en Una Guía de Onda RectangularandyÎncă nu există evaluări
- Comandos MatlabDocument3 paginiComandos MatlabManuel Augusto Alvarado HuancaÎncă nu există evaluări
- FisicaDocument25 paginiFisicaDanny Guerra EnriqueÎncă nu există evaluări
- Investigación Cafi2Document90 paginiInvestigación Cafi2Victor Nicolas Baila Oliva0% (1)
- Calculo Numerico y Mecanica Celeste PDFDocument15 paginiCalculo Numerico y Mecanica Celeste PDFcrisÎncă nu există evaluări
- Algebra de BooleDocument6 paginiAlgebra de BoolePayetCesar100% (1)
- 13 Tipos de Funciones MatemáticasDocument47 pagini13 Tipos de Funciones MatemáticasJimmy Alexander Muñoz ForeroÎncă nu există evaluări
- Ejercicios Fisica 3Document5 paginiEjercicios Fisica 3BryanÎncă nu există evaluări
- Tarea 2aDocument3 paginiTarea 2aJulissa RoblesÎncă nu există evaluări
- Cada LibreDocument3 paginiCada LibreRudy Saravia OcampoÎncă nu există evaluări
- Holder MinkowskiDocument14 paginiHolder MinkowskiJulio Cisneros LorduyÎncă nu există evaluări
- 10 Medidas - de - Dispersion - PYE PDFDocument6 pagini10 Medidas - de - Dispersion - PYE PDFLESLY NATHALIA PUPIALES RINCONÎncă nu există evaluări
- Representacion de GrafosDocument21 paginiRepresentacion de GrafosSEBASTIAN FELIPE ROJAS FRENCHÎncă nu există evaluări
- Conjuntos CompactosDocument12 paginiConjuntos CompactosRichardÎncă nu există evaluări
- Ev SiDocument15 paginiEv Siclaso87Încă nu există evaluări
- RPT Doc HRManualDocument2 paginiRPT Doc HRManualJhon ArteagaÎncă nu există evaluări
- Moriello - Dinamica de Los Sistemas ComplejosDocument9 paginiMoriello - Dinamica de Los Sistemas ComplejosJuan Carlos PinedaÎncă nu există evaluări
- Cloudcomputing 10s01Document9 paginiCloudcomputing 10s01Edson Ugarte LauraÎncă nu există evaluări
- Los Delitos InformxticosDocument20 paginiLos Delitos InformxticosN C AÎncă nu există evaluări
- Para La Verificación de Que Los Niveles de RadiaciónDocument12 paginiPara La Verificación de Que Los Niveles de RadiaciónJhon ArteagaÎncă nu există evaluări
- Ley Especial Contra Delitos InformáticosDocument7 paginiLey Especial Contra Delitos Informáticosseperezg100% (1)
- Algoritmos ProgramacionDocument96 paginiAlgoritmos Programacionvalita23Încă nu există evaluări
- DisenobdDocument3 paginiDisenobdJeanMartin CaviedesÎncă nu există evaluări
- Sistema de Recomendacion para RetailDocument79 paginiSistema de Recomendacion para RetailJhon ArteagaÎncă nu există evaluări
- Practicas ProjectDocument16 paginiPracticas ProjectCarmelo100% (6)
- Tipos MysqlDocument2 paginiTipos MysqlJhon ArteagaÎncă nu există evaluări
- Curso Basico ProjectDocument25 paginiCurso Basico ProjectGeorge RamosÎncă nu există evaluări
- Ejercicio de Access 2003Document10 paginiEjercicio de Access 2003Daniel Peña SaenzÎncă nu există evaluări
- Algoritmo KNNDocument8 paginiAlgoritmo KNNAlexis Sangoquiza AvilésÎncă nu există evaluări
- KNN TiposDocument4 paginiKNN TiposJhon ArteagaÎncă nu există evaluări
- KNN Basado en DisimilitudesDocument44 paginiKNN Basado en DisimilitudesJhon ArteagaÎncă nu există evaluări
- Capitulo I - AccessDocument68 paginiCapitulo I - AccessJhon ArteagaÎncă nu există evaluări
- Diagramas ERRDocument3 paginiDiagramas ERRJhon ArteagaÎncă nu există evaluări
- Funciones de ExcelDocument8 paginiFunciones de ExcelJhon ArteagaÎncă nu există evaluări
- 15actitudesparaelfracaso Bernardostamateas 110116155101 Phpapp01.ppsDocument16 pagini15actitudesparaelfracaso Bernardostamateas 110116155101 Phpapp01.ppsJhon ArteagaÎncă nu există evaluări
- Manual TramitesDocument162 paginiManual Tramitesjuan carlosÎncă nu există evaluări
- Ruta CríticaDocument23 paginiRuta CríticaManuel Borrego93% (14)
- Elcuervoedgarallanpoe 110729191235 Phpapp01Document8 paginiElcuervoedgarallanpoe 110729191235 Phpapp01Jhon ArteagaÎncă nu există evaluări
- Universidad Privada Domingo SavioDocument18 paginiUniversidad Privada Domingo SavioJhon ArteagaÎncă nu există evaluări
- 15actitudesparaelfracaso Bernardostamateas 110116155101 Phpapp01.ppsDocument16 pagini15actitudesparaelfracaso Bernardostamateas 110116155101 Phpapp01.ppsJhon ArteagaÎncă nu există evaluări
- Module c1Document67 paginiModule c1Liam JJÎncă nu există evaluări
- Métodos y Funciones en Java PDFDocument4 paginiMétodos y Funciones en Java PDFWillian Almanza VallejoÎncă nu există evaluări
- Ada95 PDFDocument48 paginiAda95 PDFggonaldÎncă nu există evaluări
- Hoja de Ruta - Desarrollo WebDocument27 paginiHoja de Ruta - Desarrollo Webagustin lopezÎncă nu există evaluări
- Lenguaje Estructurado de Consultas SQL CompressDocument33 paginiLenguaje Estructurado de Consultas SQL Compressjoel jose ascanio rodrigues100% (1)
- 1-Simulacion Codigos de LineaDocument4 pagini1-Simulacion Codigos de LineaJuliana CantilloÎncă nu există evaluări
- Manual JAVA y Ejercicio PDFDocument98 paginiManual JAVA y Ejercicio PDFyensis50% (2)
- Administracion de FrutaDocument4 paginiAdministracion de Frutadiegocantemateo5Încă nu există evaluări
- Curso Avanzado de Matlab 7Document27 paginiCurso Avanzado de Matlab 7anafabizÎncă nu există evaluări
- Com Vendedores WhileDocument3 paginiCom Vendedores WhileLiliana ReyesÎncă nu există evaluări
- Comandos Dos Windows XPDocument3 paginiComandos Dos Windows XPpjaironÎncă nu există evaluări
- Activ. No 1 AlgoritDocument7 paginiActiv. No 1 AlgoritDay Fu100% (1)
- Informe de Laboratorio Cobertura de Código: Fernandez Lopez Luciana AlexandraDocument2 paginiInforme de Laboratorio Cobertura de Código: Fernandez Lopez Luciana AlexandraAndrea FernandezÎncă nu există evaluări
- Control Semana 2 Ariel Vergara AvalosDocument10 paginiControl Semana 2 Ariel Vergara AvalosJULIO VERGARA MEDINAÎncă nu există evaluări
- Optimización AbapDocument25 paginiOptimización AbappacoÎncă nu există evaluări
- Capítulo 8. Libro Davini M.C.Document7 paginiCapítulo 8. Libro Davini M.C.Valentina KiefferÎncă nu există evaluări
- Manual de ABAP by MundosapDocument47 paginiManual de ABAP by MundosapJulio RafaelÎncă nu există evaluări
- PRÁCTICA 1 - BrunoErickFríasReséndizDocument62 paginiPRÁCTICA 1 - BrunoErickFríasReséndizBruno friasÎncă nu există evaluări
- Bomba ForkDocument10 paginiBomba ForkMaya GerardoÎncă nu există evaluări
- Cómo Hacer Una Calculadora en Visual BasicDocument10 paginiCómo Hacer Una Calculadora en Visual BasicAmarelis YbarraÎncă nu există evaluări
- Método Simplex Investigación de OperacionesDocument9 paginiMétodo Simplex Investigación de OperacionesEduardo PerezÎncă nu există evaluări
- Informatica Aplicada A La Educacion. NOM PDFDocument2 paginiInformatica Aplicada A La Educacion. NOM PDFJavier AlcivarÎncă nu există evaluări
- Dossier de Prensa. Kerube González 25I107Document9 paginiDossier de Prensa. Kerube González 25I107Kerube MabelÎncă nu există evaluări
- 09 - Abb (BST)Document25 pagini09 - Abb (BST)a01663882Încă nu există evaluări
- Origen de PythonDocument3 paginiOrigen de PythonAnthony Zenteno HernandezÎncă nu există evaluări
- Teoria de Matematicas Discretas 2 para La UTDocument2 paginiTeoria de Matematicas Discretas 2 para La UTJhonattan Villamil ZamoraÎncă nu există evaluări
- Modalidad de Exámenes - Semana 4 - Revisión Del IntentoDocument3 paginiModalidad de Exámenes - Semana 4 - Revisión Del IntentoJuan Jose Ruiz100% (4)
- Resumen Sesiones 1 y 2 EPEDDocument9 paginiResumen Sesiones 1 y 2 EPEDachilipunÎncă nu există evaluări
- Quiz 2 - Semana 7 - Ra - Segundo Bloque-Analisis y Verificacion de Algoritmos - (Grupo1) Intento 1Document7 paginiQuiz 2 - Semana 7 - Ra - Segundo Bloque-Analisis y Verificacion de Algoritmos - (Grupo1) Intento 1Liliana Amaya SalcedoÎncă nu există evaluări
- C (Autoguardado)Document2 paginiC (Autoguardado)Maicol GordilloÎncă nu există evaluări