S-ar putea să vă placă și
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (74)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- Csm6 Ext1y11 BookDocument955 paginiCsm6 Ext1y11 BookJesse Davis100% (12)
- Gigold PDFDocument61 paginiGigold PDFSurender SinghÎncă nu există evaluări
- Habanera Botolena & Carinosa (Gas-A)Document8 paginiHabanera Botolena & Carinosa (Gas-A)christian100% (4)
- Previous SlideDocument1 paginăPrevious SlideGupta Prem ShankarÎncă nu există evaluări
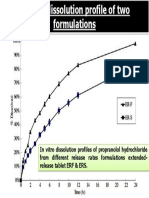
- In Vitro Dissolution Profiles of Propranolol Hydrochloride From Different Release Rates Formulations Extended-Release Tablet ERF & ERSDocument1 paginăIn Vitro Dissolution Profiles of Propranolol Hydrochloride From Different Release Rates Formulations Extended-Release Tablet ERF & ERSGupta Prem ShankarÎncă nu există evaluări
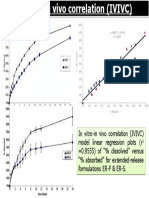
- IVIVCDocument1 paginăIVIVCGupta Prem ShankarÎncă nu există evaluări
- Various ArticlesDocument6 paginiVarious ArticlesGupta Prem ShankarÎncă nu există evaluări
- References For ChapterDocument4 paginiReferences For ChapterGupta Prem ShankarÎncă nu există evaluări
- Microfinance and Economic Empowerment of WomenDocument17 paginiMicrofinance and Economic Empowerment of WomenNamita GuptaÎncă nu există evaluări
- Mycology Chapter 2v2Document10 paginiMycology Chapter 2v2abatabrahamÎncă nu există evaluări
- PremDocument35 paginiPremGupta Prem ShankarÎncă nu există evaluări
- Institute of Actuaries of India: Subject CT3-Probability and Mathematical Statistics May 2008 ExaminationDocument10 paginiInstitute of Actuaries of India: Subject CT3-Probability and Mathematical Statistics May 2008 ExaminationeuticusÎncă nu există evaluări
- Life in The Past - Year 6 WorksheetsDocument11 paginiLife in The Past - Year 6 WorksheetstinaÎncă nu există evaluări
- Lecture 6Document7 paginiLecture 6Shuja MirÎncă nu există evaluări
- Sta. Lucia National High School: Republic of The Philippines Region III-Central LuzonDocument7 paginiSta. Lucia National High School: Republic of The Philippines Region III-Central LuzonLee Charm SantosÎncă nu există evaluări
- 1sebastian Vs CalisDocument6 pagini1sebastian Vs CalisRai-chan Junior ÜÎncă nu există evaluări
- Soal Midtest + Kunci JawabanDocument28 paginiSoal Midtest + Kunci JawabanYuyun RasulongÎncă nu există evaluări
- SAP CRM Tax ConfigurationDocument18 paginiSAP CRM Tax Configurationtushar_kansaraÎncă nu există evaluări
- Strategi Pencegahan Kecelakaan Di PT VALE Indonesia Presentation To FPP Workshop - APKPI - 12102019Document35 paginiStrategi Pencegahan Kecelakaan Di PT VALE Indonesia Presentation To FPP Workshop - APKPI - 12102019Eko Maulia MahardikaÎncă nu există evaluări
- E 18 - 02 - Rte4ltay PDFDocument16 paginiE 18 - 02 - Rte4ltay PDFvinoth kumar SanthanamÎncă nu există evaluări
- Character Sketch of Elizabeth BennetDocument2 paginiCharacter Sketch of Elizabeth BennetAiman AbdullahÎncă nu există evaluări
- AmplifierDocument20 paginiAmplifierValerie StraussÎncă nu există evaluări
- Oracle QuestDocument521 paginiOracle Questprasanna ghareÎncă nu există evaluări
- Registration - No Candidate Gender Category Rank/Percentage Allotment - Seat Base - Seat Course CollegeDocument166 paginiRegistration - No Candidate Gender Category Rank/Percentage Allotment - Seat Base - Seat Course CollegeCyber ParkÎncă nu există evaluări
- RFP On Internal AuditDocument33 paginiRFP On Internal AuditCan dien tu Thai Binh DuongÎncă nu există evaluări
- 8 Powerful Methods People Use To Bounce Back From FailureDocument7 pagini8 Powerful Methods People Use To Bounce Back From FailureGrego CentillasÎncă nu există evaluări
- Impact of Micro FinanceDocument61 paginiImpact of Micro FinancePerry Arcilla SerapioÎncă nu există evaluări
- CBSE Class 12 Business Studies Question Paper 2013 With SolutionsDocument19 paginiCBSE Class 12 Business Studies Question Paper 2013 With SolutionsManormaÎncă nu există evaluări
- 3er Grado - DMPA 05 - ACTIVIDAD DE COMPRENSION LECTORA - UNIT 2 - CORRECCIONDocument11 pagini3er Grado - DMPA 05 - ACTIVIDAD DE COMPRENSION LECTORA - UNIT 2 - CORRECCIONANDERSON BRUCE MATIAS DE LA SOTAÎncă nu există evaluări
- Case - Marico SCMDocument26 paginiCase - Marico SCMChandan Gupta50% (2)
- Water On Mars PDFDocument35 paginiWater On Mars PDFAlonso GarcíaÎncă nu există evaluări
- Analysing Worship in The Pentateuch and Its ApplicationDocument12 paginiAnalysing Worship in The Pentateuch and Its ApplicationDaniel Solomon100% (1)
- Defending A Dogma: Between Grice, Strawson and Quine: Elvis ImafidonDocument10 paginiDefending A Dogma: Between Grice, Strawson and Quine: Elvis ImafidonYang Wen-LiÎncă nu există evaluări
- Ross, D. (2013) - Field Guide To Jumping Spiders of Southeast Idaho.Document4 paginiRoss, D. (2013) - Field Guide To Jumping Spiders of Southeast Idaho.Dave RossÎncă nu există evaluări
- Public Administration 2000 - CSS ForumsDocument3 paginiPublic Administration 2000 - CSS ForumsMansoor Ali KhanÎncă nu există evaluări
- 15 Melodic Uses of Non-Chord TonesDocument3 pagini15 Melodic Uses of Non-Chord TonesonlymusicaÎncă nu există evaluări
- Asset Management PlanDocument160 paginiAsset Management Planbkalatus1100% (1)
- Organigation DeveDocument3 paginiOrganigation Devemerin sunilÎncă nu există evaluări