S-ar putea să vă placă și
- NIH Public Access: Delayed Antimicrobial Therapy Increases Mortality and Organ Dysfunction Duration in Pediatric SepsisDocument23 paginiNIH Public Access: Delayed Antimicrobial Therapy Increases Mortality and Organ Dysfunction Duration in Pediatric SepsisjaneÎncă nu există evaluări
- Intratympanic Membrane Congenital CholesteatomaDocument3 paginiIntratympanic Membrane Congenital CholesteatomajaneÎncă nu există evaluări
- Radiology Icm IiDocument8 paginiRadiology Icm IijaneÎncă nu există evaluări
- By: Dr. Engelberta Sp. KJDocument14 paginiBy: Dr. Engelberta Sp. KJjaneÎncă nu există evaluări
- Substance Abuse Among Health Professionals: August 15, 2014Document25 paginiSubstance Abuse Among Health Professionals: August 15, 2014janeÎncă nu există evaluări
- Antidepressants AntidepressantsDocument38 paginiAntidepressants AntidepressantsjaneÎncă nu există evaluări
- Medical Leadership Competency Framework: Landscape of HealthcareDocument15 paginiMedical Leadership Competency Framework: Landscape of HealthcarejaneÎncă nu există evaluări
- Mechanisms of Antibiotic Resistance in BacteriaDocument40 paginiMechanisms of Antibiotic Resistance in BacteriajaneÎncă nu există evaluări
- Itching, Redness of The SkinDocument21 paginiItching, Redness of The SkinjaneÎncă nu există evaluări
- To Physiology, Homeostasis, and Body Temperature: V. Sutarmo SetiadjiDocument20 paginiTo Physiology, Homeostasis, and Body Temperature: V. Sutarmo SetiadjijaneÎncă nu există evaluări
- JawsDocument78 paginiJawsjaneÎncă nu există evaluări
- Lymphokines & Cytokines: Immunology & DiseaseDocument34 paginiLymphokines & Cytokines: Immunology & DiseasejaneÎncă nu există evaluări
- Mechanisms of Antibiotic Resistance in BacteriaDocument41 paginiMechanisms of Antibiotic Resistance in BacteriajaneÎncă nu există evaluări
- Transient Loss of Consciousness: History TakingDocument4 paginiTransient Loss of Consciousness: History TakingjaneÎncă nu există evaluări
- LibraryDocument1 paginăLibraryjaneÎncă nu există evaluări
- STM Cels Lcture 1Document44 paginiSTM Cels Lcture 1janeÎncă nu există evaluări
- COMMUNICATION With Other ProfessionalsDocument12 paginiCOMMUNICATION With Other ProfessionalsjaneÎncă nu există evaluări
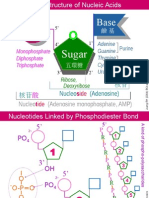
- NA01 StructureDocument5 paginiNA01 StructurejaneÎncă nu există evaluări
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (400)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (74)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (121)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- Med Sciences III Exam Ans Key-DönüştürüldüDocument8 paginiMed Sciences III Exam Ans Key-DönüştürüldüZeynep Etka ÇelikkıranÎncă nu există evaluări
- Pharmacokinetics For Test 1Document3 paginiPharmacokinetics For Test 1KaitlynÎncă nu există evaluări
- Official Chapter 2 Student Copy ZoologyDocument5 paginiOfficial Chapter 2 Student Copy Zoologyapi-528700386Încă nu există evaluări
- Biomolecules Practice QuestionsDocument4 paginiBiomolecules Practice QuestionsNoura RoseÎncă nu există evaluări
- Identifikasi Kromosom - Fauzia Rahmani - 2485597A - Kel. GDocument3 paginiIdentifikasi Kromosom - Fauzia Rahmani - 2485597A - Kel. GAstatinÎncă nu există evaluări
- Mutations Practice - KeyDocument3 paginiMutations Practice - Keyapi-263197810Încă nu există evaluări
- Fatty Acids + Physico-Chemical PropertiesDocument39 paginiFatty Acids + Physico-Chemical PropertiesSaloni AryaÎncă nu există evaluări
- Week 7-8 Photosynthesis and RespirationDocument1 paginăWeek 7-8 Photosynthesis and RespirationKenneth Roy BalangueÎncă nu există evaluări
- Ch4 FlashcardsDocument49 paginiCh4 FlashcardsShriya JagwayanÎncă nu există evaluări
- The Polycomb Group Protein Yaf2 Regulates The Pluripotency of Embryonic Stem Cells in A Phosphorylation-Dependent MannerDocument13 paginiThe Polycomb Group Protein Yaf2 Regulates The Pluripotency of Embryonic Stem Cells in A Phosphorylation-Dependent MannerSHUMETÎncă nu există evaluări
- Biochemistry of The GIT S1-10Document3 paginiBiochemistry of The GIT S1-10Kim RamosÎncă nu există evaluări
- Molecular Biology Workflow Solutions BrochureDocument56 paginiMolecular Biology Workflow Solutions BrochureBishoy F. YoussefÎncă nu există evaluări
- Cordeiro Et Al - 2015Document14 paginiCordeiro Et Al - 2015Jonas MartinsÎncă nu există evaluări
- UNSW Med Foundations Brief NotesDocument43 paginiUNSW Med Foundations Brief NotesXasdmanÎncă nu există evaluări
- The Applications of Enzymes in Industry and MedicineDocument6 paginiThe Applications of Enzymes in Industry and MedicineMary ThomasÎncă nu există evaluări
- Viruses 11 00045Document25 paginiViruses 11 00045Syarifah TasharaÎncă nu există evaluări
- Worksheet 1 Cellular AberrationDocument5 paginiWorksheet 1 Cellular AberrationKeepItSecretÎncă nu există evaluări
- Protein Chemistry-1Document38 paginiProtein Chemistry-1Janhvi100% (1)
- Atherosclerosis: Reporter: Collera, Charissa Constantino, Venice Clemena, Adnan Coronel, Romeo Cordova, KarlaDocument73 paginiAtherosclerosis: Reporter: Collera, Charissa Constantino, Venice Clemena, Adnan Coronel, Romeo Cordova, Karlaprecious_bustosÎncă nu există evaluări
- Final Neet (Ug) - 2020 Examination: Biology Test Paper With AnswerDocument10 paginiFinal Neet (Ug) - 2020 Examination: Biology Test Paper With AnswerL V Laxmipathi RaoÎncă nu există evaluări
- PHG 413 - Vitamin C b1 b2 b3Document56 paginiPHG 413 - Vitamin C b1 b2 b3Indra punya ProjectÎncă nu există evaluări
- General Biology 1 Quarter 1 Week 2.1: CapsletDocument7 paginiGeneral Biology 1 Quarter 1 Week 2.1: CapsletAmil, Shierly Mae S. -10 QUISUMBINGÎncă nu există evaluări
- 2 PHAR0004 9 7 ReceptorStructure NSM 2018 NotesDocument3 pagini2 PHAR0004 9 7 ReceptorStructure NSM 2018 NotesArthi ArumukasamyÎncă nu există evaluări
- MatrizDocument91 paginiMatrizndsjÎncă nu există evaluări
- Prokaryotic&Eukaryotic DIFFERENCESDocument7 paginiProkaryotic&Eukaryotic DIFFERENCESChristian John Sitjar Dumo100% (1)
- Polymerase Chain Reac6on PCR: What To Do Today?Document7 paginiPolymerase Chain Reac6on PCR: What To Do Today?Juliana MaltaÎncă nu există evaluări
- PPTDocument30 paginiPPTAimanÎncă nu există evaluări
- BioPhotons DNADocument2 paginiBioPhotons DNAΜακης ΣακεταςÎncă nu există evaluări
- Lonza BenchGuides SourceBook Section VII - Separation of DNA in Polyacrylamide GelsDocument6 paginiLonza BenchGuides SourceBook Section VII - Separation of DNA in Polyacrylamide GelsOsama AbdulkareemÎncă nu există evaluări
- Biological Value of Protein Rich FoodsDocument4 paginiBiological Value of Protein Rich FoodsMaryam ShahzadiÎncă nu există evaluări