S-ar putea să vă placă și
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDe la EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryEvaluare: 3.5 din 5 stele3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)De la EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Evaluare: 4.5 din 5 stele4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDe la EverandNever Split the Difference: Negotiating As If Your Life Depended On ItEvaluare: 4.5 din 5 stele4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDe la EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaEvaluare: 4.5 din 5 stele4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingDe la EverandThe Little Book of Hygge: Danish Secrets to Happy LivingEvaluare: 3.5 din 5 stele3.5/5 (399)
- Grit: The Power of Passion and PerseveranceDe la EverandGrit: The Power of Passion and PerseveranceEvaluare: 4 din 5 stele4/5 (587)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDe la EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyEvaluare: 3.5 din 5 stele3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDe la EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeEvaluare: 4 din 5 stele4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnDe la EverandTeam of Rivals: The Political Genius of Abraham LincolnEvaluare: 4.5 din 5 stele4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeDe la EverandShoe Dog: A Memoir by the Creator of NikeEvaluare: 4.5 din 5 stele4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDe la EverandThe Emperor of All Maladies: A Biography of CancerEvaluare: 4.5 din 5 stele4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDe la EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreEvaluare: 4 din 5 stele4/5 (1090)
- Her Body and Other Parties: StoriesDe la EverandHer Body and Other Parties: StoriesEvaluare: 4 din 5 stele4/5 (821)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDe la EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersEvaluare: 4.5 din 5 stele4.5/5 (344)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDe la EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceEvaluare: 4 din 5 stele4/5 (890)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDe la EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureEvaluare: 4.5 din 5 stele4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaDe la EverandThe Unwinding: An Inner History of the New AmericaEvaluare: 4 din 5 stele4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)De la EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Evaluare: 4 din 5 stele4/5 (98)
- On Fire: The (Burning) Case for a Green New DealDe la EverandOn Fire: The (Burning) Case for a Green New DealEvaluare: 4 din 5 stele4/5 (73)
- Ledger - Problems and SolutionsDocument1 paginăLedger - Problems and SolutionsDjamal SalimÎncă nu există evaluări
- Theoritical and Applied LinguisticDocument6 paginiTheoritical and Applied LinguisticOdonkz Forrealracingtiga100% (2)
- Unitized Curtain Wall SystemDocument38 paginiUnitized Curtain Wall Systems.senthil nathan100% (1)
- Fluorescence-Activated Cell SortingDocument10 paginiFluorescence-Activated Cell SortingShaher Bano MirzaÎncă nu există evaluări
- Reference Guide for Pineapple JuiceDocument5 paginiReference Guide for Pineapple JuiceLayfloÎncă nu există evaluări
- VFTO DocumentationDocument119 paginiVFTO DocumentationSheri Abhishek ReddyÎncă nu există evaluări
- Cytoscape3 7 0manual PDFDocument249 paginiCytoscape3 7 0manual PDFHalil İbrahim ÖzdemirÎncă nu există evaluări
- Bibliography PDFDocument20 paginiBibliography PDFHalil İbrahim ÖzdemirÎncă nu există evaluări
- Identifying HER2 Inhibitors From Natural Products Database PDFDocument9 paginiIdentifying HER2 Inhibitors From Natural Products Database PDFHalil İbrahim ÖzdemirÎncă nu există evaluări
- Preface: 1 2 Nonequilibrium Thermodynamics and Continuum MechanicsDocument4 paginiPreface: 1 2 Nonequilibrium Thermodynamics and Continuum MechanicsHalil İbrahim ÖzdemirÎncă nu există evaluări
- IndexDocument5 paginiIndexHalil İbrahim ÖzdemirÎncă nu există evaluări
- Frontmatter PDFDocument4 paginiFrontmatter PDFHalil İbrahim ÖzdemirÎncă nu există evaluări
- Bibliography PDFDocument20 paginiBibliography PDFHalil İbrahim ÖzdemirÎncă nu există evaluări
- IndexDocument5 paginiIndexHalil İbrahim ÖzdemirÎncă nu există evaluări
- Visualizing Markov models for biomolecule foldingDocument3 paginiVisualizing Markov models for biomolecule foldingHalil İbrahim ÖzdemirÎncă nu există evaluări
- Preface: 1 2 Nonequilibrium Thermodynamics and Continuum MechanicsDocument4 paginiPreface: 1 2 Nonequilibrium Thermodynamics and Continuum MechanicsHalil İbrahim ÖzdemirÎncă nu există evaluări
- PrefaceDocument2 paginiPrefaceHalil İbrahim ÖzdemirÎncă nu există evaluări
- Visualizing Markov models for biomolecule foldingDocument3 paginiVisualizing Markov models for biomolecule foldingHalil İbrahim ÖzdemirÎncă nu există evaluări
- CHE 379/CHE384: Biochemical, Cellular, and Metabolic Engineering: Principles and PracticesDocument4 paginiCHE 379/CHE384: Biochemical, Cellular, and Metabolic Engineering: Principles and PracticesHalil İbrahim ÖzdemirÎncă nu există evaluări
- PrefaceDocument2 paginiPrefaceHalil İbrahim ÖzdemirÎncă nu există evaluări
- Target IdentificationDocument6 paginiTarget IdentificationHalil İbrahim ÖzdemirÎncă nu există evaluări
- Lecture5 PpsDocument29 paginiLecture5 PpsHalil İbrahim ÖzdemirÎncă nu există evaluări
- Final GBDocument10 paginiFinal GBHalil İbrahim ÖzdemirÎncă nu există evaluări
- Produce BioethanolDocument11 paginiProduce BioethanolHalil İbrahim ÖzdemirÎncă nu există evaluări
- Bio StringsDocument154 paginiBio StringsHalil İbrahim ÖzdemirÎncă nu există evaluări
- Study View Clinical DataDocument80 paginiStudy View Clinical DataHalil İbrahim ÖzdemirÎncă nu există evaluări
- Hesaplamalarda KullanDocument26 paginiHesaplamalarda KullanHalil İbrahim ÖzdemirÎncă nu există evaluări
- Material BalancesDocument15 paginiMaterial BalancesHalil İbrahim ÖzdemirÎncă nu există evaluări
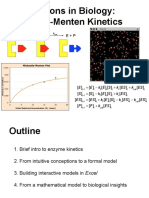
- Michaelis-Menten Talk - FullDocument18 paginiMichaelis-Menten Talk - FullHalil İbrahim ÖzdemirÎncă nu există evaluări
- LUTS Spot TestDocument2 paginiLUTS Spot TestHardiTariqHamma100% (1)
- DHRM 3 SyllabusDocument10 paginiDHRM 3 SyllabusCherokee Tuazon RodriguezÎncă nu există evaluări
- Duct Seal: Multi Cable and Pipe Sealing SystemDocument8 paginiDuct Seal: Multi Cable and Pipe Sealing SystemSri VarshiniÎncă nu există evaluări
- Essay #01 (First Draft)Document2 paginiEssay #01 (First Draft)thanhtam3819Încă nu există evaluări
- Rabuddha HarataDocument67 paginiRabuddha HaratasagggasgfaÎncă nu există evaluări
- Parameter Pengelasan SMAW: No Bahan Diameter Ampere Polaritas Penetrasi Rekomendasi Posisi PengguanaanDocument2 paginiParameter Pengelasan SMAW: No Bahan Diameter Ampere Polaritas Penetrasi Rekomendasi Posisi PengguanaanKhamdi AfandiÎncă nu există evaluări
- Metaswitch Datasheet Network Transformation OverviewDocument5 paginiMetaswitch Datasheet Network Transformation OverviewblitoÎncă nu există evaluări
- Datasheet PIC1650Document7 paginiDatasheet PIC1650Vinicius BaconÎncă nu există evaluări
- The DHCP Snooping and DHCP Alert Method in SecurinDocument9 paginiThe DHCP Snooping and DHCP Alert Method in SecurinSouihi IslemÎncă nu există evaluări
- Key-Words: - Techniques, Reflection, Corporal Punishment, EffectiveDocument7 paginiKey-Words: - Techniques, Reflection, Corporal Punishment, EffectiveManawÎncă nu există evaluări
- Production of Carotenoids From Rhodotorula Mucilaginosa and Their Applications As Colorant Agent in Sweet CandyDocument7 paginiProduction of Carotenoids From Rhodotorula Mucilaginosa and Their Applications As Colorant Agent in Sweet CandyEspinosa Balderas GenaroÎncă nu există evaluări
- ME 205 - Statics Course Syllabus: Fall 2015Document4 paginiME 205 - Statics Course Syllabus: Fall 2015Dhenil ManubatÎncă nu există evaluări
- Anselm's Ontological Argument ExplainedDocument8 paginiAnselm's Ontological Argument ExplainedCharles NunezÎncă nu există evaluări
- Land, Soil, Water, Natural Vegetation& Wildlife ResourcesDocument26 paginiLand, Soil, Water, Natural Vegetation& Wildlife ResourcesKritika VermaÎncă nu există evaluări
- Renold Transmission Chain Catalogue ENG 0112Document94 paginiRenold Transmission Chain Catalogue ENG 0112nataliaÎncă nu există evaluări
- Excel File - Business Computing - End Term (To Be Shared Vide Email)Document301 paginiExcel File - Business Computing - End Term (To Be Shared Vide Email)Aman SankrityayanÎncă nu există evaluări
- Chapter #11: Volume #1Document35 paginiChapter #11: Volume #1Mohamed MohamedÎncă nu există evaluări
- TK17 V10 ReadmeDocument72 paginiTK17 V10 ReadmePaula PérezÎncă nu există evaluări
- Cpar Lesson 1Document44 paginiCpar Lesson 1althea villanuevaÎncă nu există evaluări
- Handout 4-6 StratDocument6 paginiHandout 4-6 StratTrixie JordanÎncă nu există evaluări
- Using The 12c Real-Time SQL Monitoring Report History For Performance AnalysisDocument39 paginiUsing The 12c Real-Time SQL Monitoring Report History For Performance AnalysisJack WangÎncă nu există evaluări
- 4.6.6 Lab View Wired and Wireless Nic InformationDocument4 pagini4.6.6 Lab View Wired and Wireless Nic InformationThắng NguyễnÎncă nu există evaluări
- Total Cost of Ownership in PV Manufacturing - GuideDocument12 paginiTotal Cost of Ownership in PV Manufacturing - GuidebirlainÎncă nu există evaluări
- Alcatel 350 User Guide FeaturesDocument4 paginiAlcatel 350 User Guide FeaturesFilipe CardosoÎncă nu există evaluări
- UI Symphony Orchestra and Choirs Concert Features Mahler's ResurrectionDocument17 paginiUI Symphony Orchestra and Choirs Concert Features Mahler's ResurrectionJilly CookeÎncă nu există evaluări