S-ar putea să vă placă și
- Alimente care ucid creierul. Cerealele, zahărul și alți carbohidrațiDe la EverandAlimente care ucid creierul. Cerealele, zahărul și alți carbohidrațiEvaluare: 5 din 5 stele5/5 (4)
- In abis: Insemnarile unui neuropsihiatru despre tulburarile mintiiDe la EverandIn abis: Insemnarile unui neuropsihiatru despre tulburarile mintiiÎncă nu există evaluări
- Dieta pentru zile senine II: De la durere la echilibru spiritual, psihologic, nutritivDe la EverandDieta pentru zile senine II: De la durere la echilibru spiritual, psihologic, nutritivEvaluare: 5 din 5 stele5/5 (1)
- Bolile nu apar chiar din senin: Impactul evenimentelor negative asupra sănătății noastreDe la EverandBolile nu apar chiar din senin: Impactul evenimentelor negative asupra sănătății noastreÎncă nu există evaluări
- Maladii HeterozomaleDocument4 paginiMaladii HeterozomaleJustyn Cristian100% (1)
- Sindromul DownDocument8 paginiSindromul DownDorian Mihai Ciubotaru100% (1)
- Boli GeneticeDocument68 paginiBoli GeneticeElena Pîrciu100% (2)
- WWW - Referate.ro-Sindromul Turner c5f48Document3 paginiWWW - Referate.ro-Sindromul Turner c5f48Evelina RosuÎncă nu există evaluări
- AneuploidiaDocument7 paginiAneuploidiamagdaÎncă nu există evaluări
- Curs 6Document75 paginiCurs 6OanaÎncă nu există evaluări
- Sindrom Down Referat 3Document3 paginiSindrom Down Referat 3Rada LiviaÎncă nu există evaluări
- Sindromul Turner. ModifDocument14 paginiSindromul Turner. ModifabcÎncă nu există evaluări
- Sindroame CromozomialeDocument106 paginiSindroame CromozomialeAndreea NeculceaÎncă nu există evaluări
- Curs 9 MG Bolile CromosomiceDocument54 paginiCurs 9 MG Bolile CromosomiceCristina DorofteÎncă nu există evaluări
- Sindromul Wolf-HirschhornDocument12 paginiSindromul Wolf-HirschhornMarian Fotescu100% (1)
- Referat Embriogenetica Sindromul PatauDocument15 paginiReferat Embriogenetica Sindromul PatauAnonymous oiyzQqTWT4Încă nu există evaluări
- Fiziologia RenalaDocument11 paginiFiziologia RenalaIoana Tudor100% (1)
- Sindromul TurnerDocument8 paginiSindromul Turnerhadad_salma_48088144100% (1)
- Trisomia 21Document8 paginiTrisomia 21AncaÎncă nu există evaluări
- Referat - Boli CromozomialeDocument6 paginiReferat - Boli CromozomialeCorina ȘerbănescuÎncă nu există evaluări
- Sindrom DownDocument5 paginiSindrom DownDaniela Rusnac100% (1)
- Maladiile GeneticeDocument37 paginiMaladiile GeneticeAlexia Oproiu100% (2)
- Maladii EreditareDocument25 paginiMaladii EreditareAnastasia RusuÎncă nu există evaluări
- Bolile Cromozomiale Pentru StudentiDocument78 paginiBolile Cromozomiale Pentru StudentiCristina Porfire100% (1)
- Anomaliile CromozomialeDocument6 paginiAnomaliile CromozomialeMihai VidreanÎncă nu există evaluări
- LP Sindroame Cromozomiale - Cazuri Aplicate RoxanaDocument10 paginiLP Sindroame Cromozomiale - Cazuri Aplicate RoxanaIulian IulianÎncă nu există evaluări
- Diagnosticul GeneticDocument56 paginiDiagnosticul GeneticAndreea CiubotaruÎncă nu există evaluări
- Bolile CromozomialeDocument8 paginiBolile CromozomialeFlorina PaulaÎncă nu există evaluări
- Sindroame AutozomiceDocument6 paginiSindroame AutozomicePopescuRamonaÎncă nu există evaluări
- Poliploidia Si Aneuploidia 4Document61 paginiPoliploidia Si Aneuploidia 4UN Q DeeÎncă nu există evaluări
- Maladii CromozomialeDocument6 paginiMaladii CromozomialeNicoleta FerecatuÎncă nu există evaluări
- Anomalii de NumarDocument31 paginiAnomalii de NumarRares MarinÎncă nu există evaluări
- Genetică Umană, Boli GeneticeDocument2 paginiGenetică Umană, Boli GeneticeAlex PrunaÎncă nu există evaluări
- Genetica LP EXAMENDocument10 paginiGenetica LP EXAMENirrrsÎncă nu există evaluări
- Trisomia PatauDocument3 paginiTrisomia PatauViCky MicKyÎncă nu există evaluări
- Anomaliile CromozomialeDocument20 paginiAnomaliile CromozomialePopescu DanielÎncă nu există evaluări
- Boli GeneticeDocument12 paginiBoli Geneticegeorgyana_pÎncă nu există evaluări
- Anomaliile Cromozomiale Umane Şi Efectele Lor FenotipiceDocument8 paginiAnomaliile Cromozomiale Umane Şi Efectele Lor FenotipiceAlexandra ViolettÎncă nu există evaluări
- Boli GeneticeDocument9 paginiBoli GeneticeLaila MdalalÎncă nu există evaluări
- Afectiuni GeneticeDocument15 paginiAfectiuni GeneticeSadDaniela 13Încă nu există evaluări
- Indicatii CariotipDocument16 paginiIndicatii CariotipAristotle L.Încă nu există evaluări
- Curs Boli CromozomialeDocument53 paginiCurs Boli Cromozomialeteodor_cristea1947Încă nu există evaluări
- Genetica 2.0Document5 paginiGenetica 2.0Alexia TuțescuÎncă nu există evaluări
- SindroameDocument10 paginiSindroamefrancescalaslau32Încă nu există evaluări
- Maladii Produse de Mutaţii GenomiceDocument19 paginiMaladii Produse de Mutaţii GenomiceSzabo RamonaÎncă nu există evaluări
- Sindroame Genetice-CaracteristiciDocument17 paginiSindroame Genetice-CaracteristiciAlexandra TițaÎncă nu există evaluări
- Referat BiologieDocument7 paginiReferat BiologieRobertGlințăÎncă nu există evaluări
- Sindr DownDocument6 paginiSindr DownClaudia IonitaÎncă nu există evaluări
- Bolile CromozomialeDocument4 paginiBolile Cromozomiale•《 Adela 》•Încă nu există evaluări
- Proiect Genetica Boli Produse de Anomalii CromozomialeDocument54 paginiProiect Genetica Boli Produse de Anomalii CromozomialeIstrate AlexandraÎncă nu există evaluări
- Prezentare 9Document6 paginiPrezentare 9brunoÎncă nu există evaluări
- Maladii Cromozomiale Autozomale ProiectDocument35 paginiMaladii Cromozomiale Autozomale ProiectCozminel MaimutelÎncă nu există evaluări
- Sindroame GeneticeDocument9 paginiSindroame GeneticeFlorentina GmnrÎncă nu există evaluări
- Curs 9 MG Bolile CromosomiceDocument54 paginiCurs 9 MG Bolile CromosomiceDiana IacobÎncă nu există evaluări
- Boli Genetice Si de MetabolismDocument72 paginiBoli Genetice Si de MetabolismCosmin CozmaÎncă nu există evaluări
- Maladii HeterozomaleDocument3 paginiMaladii Heterozomaleayu apriliaÎncă nu există evaluări
- Sindroame cz4Document26 paginiSindroame cz4DemnitateÎncă nu există evaluări
- Anomalii CromozomialeDocument11 paginiAnomalii CromozomialeGeorge VladÎncă nu există evaluări
- Malformatii CongenitaleDocument5 paginiMalformatii CongenitaleAlina Camerzan50% (2)
- Maladii GeneticeDocument5 paginiMaladii GeneticeTudor ZapciroiuÎncă nu există evaluări
- Tromboangeita Obliterantă - Boala BuergerDocument7 paginiTromboangeita Obliterantă - Boala BuergerIoana TudorÎncă nu există evaluări
- Chiru 4Document6 paginiChiru 4Ioana TudorÎncă nu există evaluări
- CURS NR 4 GeneticaDocument7 paginiCURS NR 4 GeneticaIoana TudorÎncă nu există evaluări
- Chiru 2Document6 paginiChiru 2Ioana TudorÎncă nu există evaluări
- EXPLORAREA ACUITĂȚII VIZUALE - Tudor IoanaDocument6 paginiEXPLORAREA ACUITĂȚII VIZUALE - Tudor IoanaIoana TudorÎncă nu există evaluări
- Statia 2 - Monitorizarea PacientuluiDocument2 paginiStatia 2 - Monitorizarea PacientuluiIoana TudorÎncă nu există evaluări
- Examen ASPDocument4 paginiExamen ASPIoana TudorÎncă nu există evaluări
- Relatia Medic+ PacientDocument8 paginiRelatia Medic+ PacientIoana TudorÎncă nu există evaluări
- Digestia mg1-1Document54 paginiDigestia mg1-1Ioana Tudor100% (1)
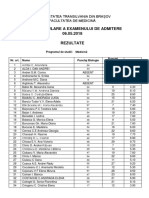
- Rezultate Simulare 2018 - MD Si MDADocument5 paginiRezultate Simulare 2018 - MD Si MDAIoana TudorÎncă nu există evaluări